

Phylogenetic background and habitat drive the genetic diversification of Escherichia coli
- PMID: 32530914
- PMCID: PMC7314097
- DOI: 10.1371/journal.pgen.1008866
Phylogenetic background and habitat drive the genetic diversification of Escherichia coli
Abstract
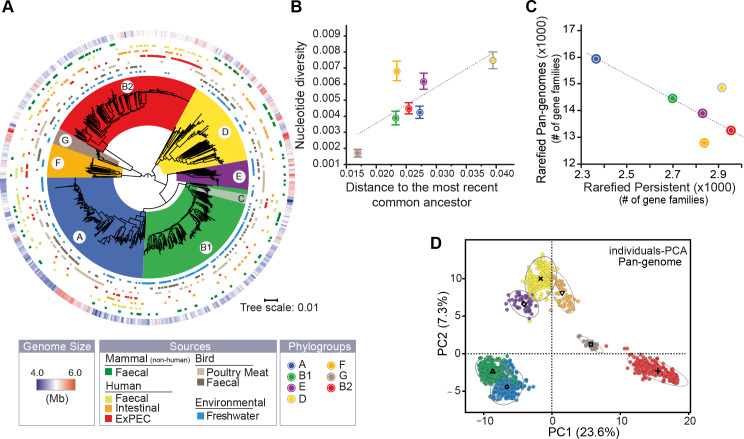
Escherichia coli is mostly a commensal of birds and mammals, including humans, where it can act as an opportunistic pathogen. It is also found in water and sediments. We investigated the phylogeny, genetic diversification, and habitat-association of 1,294 isolates representative of the phylogenetic diversity of more than 5,000 isolates from the Australian continent. Since many previous studies focused on clinical isolates, we investigated mostly other isolates originating from humans, poultry, wild animals and water. These strains represent the species genetic diversity and reveal widespread associations between phylogroups and isolation sources. The analysis of strains from the same sequence types revealed very rapid change of gene repertoires in the very early stages of divergence, driven by the acquisition of many different types of mobile genetic elements. These elements also lead to rapid variations in genome size, even if few of their genes rise to high frequency in the species. Variations in genome size are associated with phylogroup and isolation sources, but the latter determine the number of MGEs, a marker of recent transfer, suggesting that gene flow reinforces the association of certain genetic backgrounds with specific habitats. After a while, the divergence of gene repertoires becomes linear with phylogenetic distance, presumably reflecting the continuous turnover of mobile element and the occasional acquisition of adaptive genes. Surprisingly, the phylogroups with smallest genomes have the highest rates of gene repertoire diversification and fewer but more diverse mobile genetic elements. This suggests that smaller genomes are associated with higher, not lower, turnover of genetic information. Many of these genomes are from freshwater isolates and have peculiar traits, including a specific capsule, suggesting adaptation to this environment. Altogether, these data contribute to explain why epidemiological clones tend to emerge from specific phylogenetic groups in the presence of pervasive horizontal gene transfer across the species.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures







References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
