

FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds
- PMID: 32531203
- PMCID: PMC7415647
- DOI: 10.1016/j.str.2020.05.011
FARFAR2: Improved De Novo Rosetta Prediction of Complex Global RNA Folds
Abstract
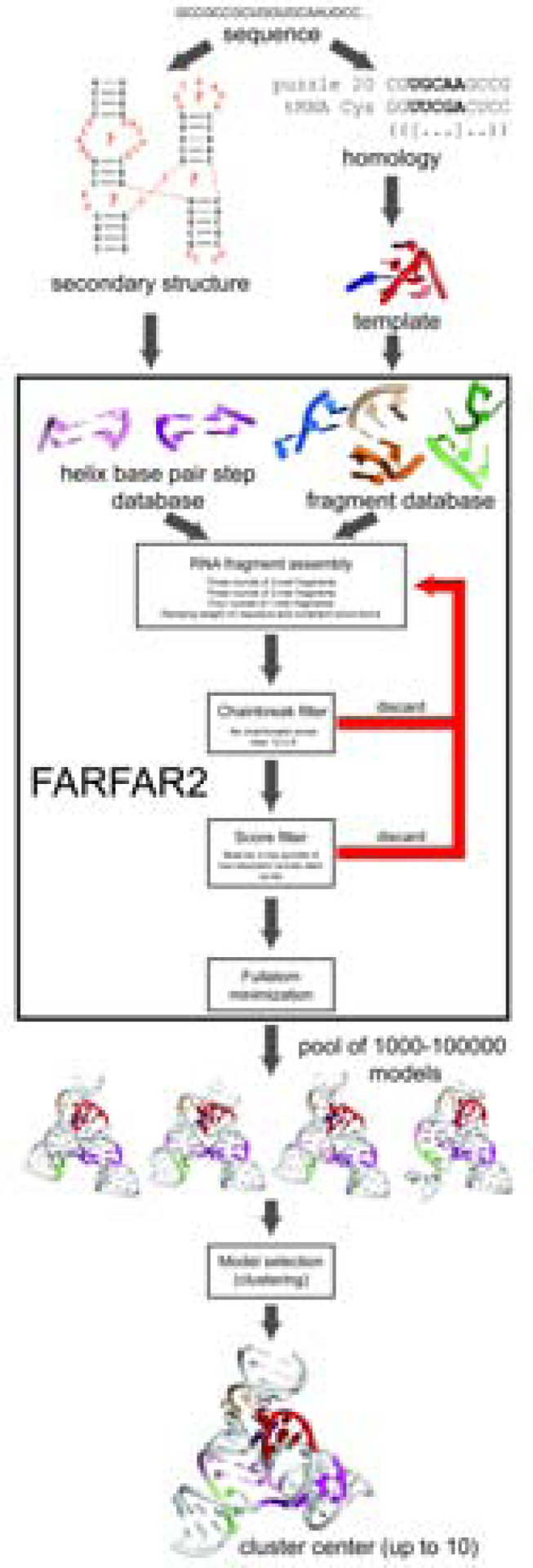
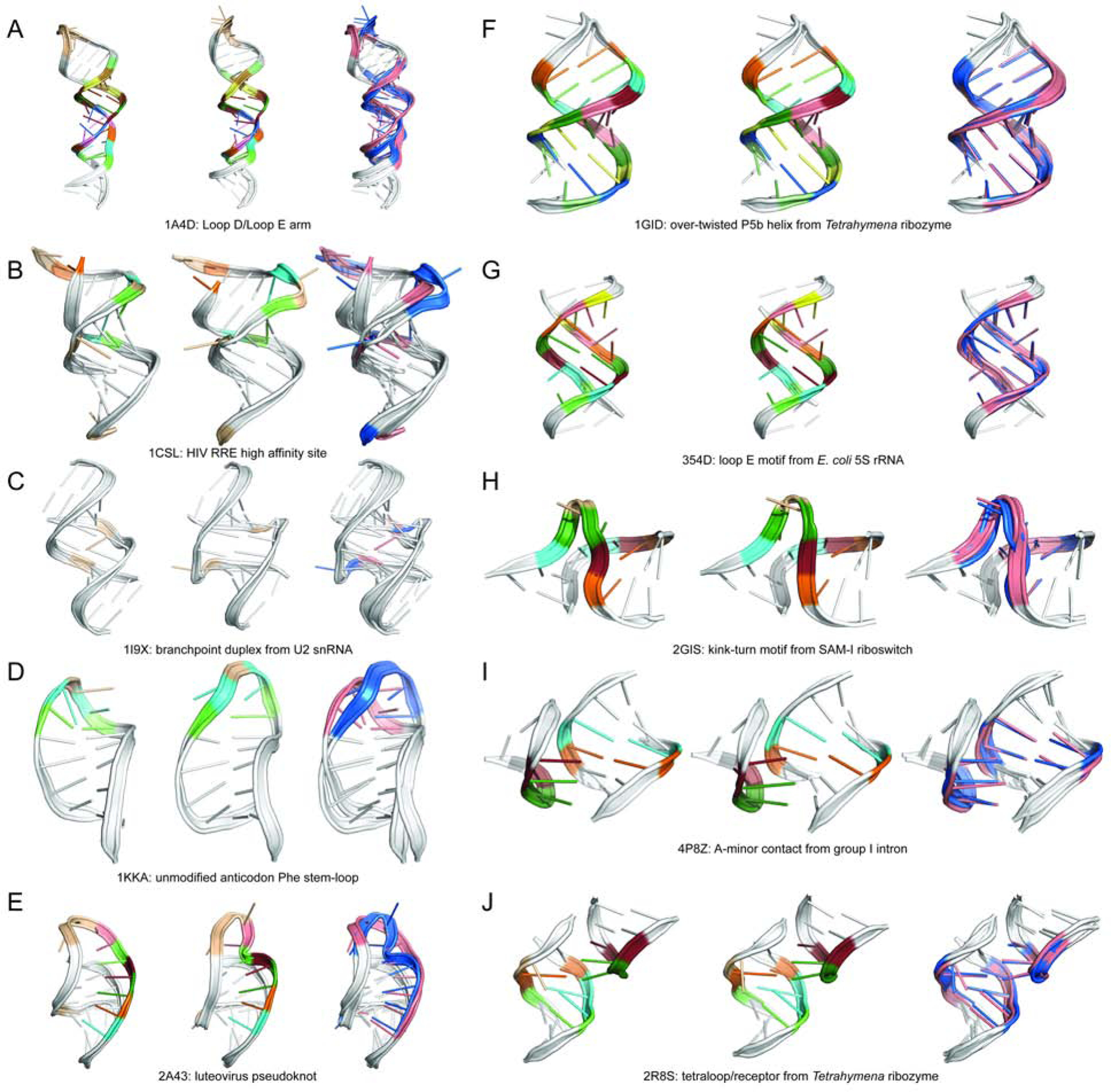
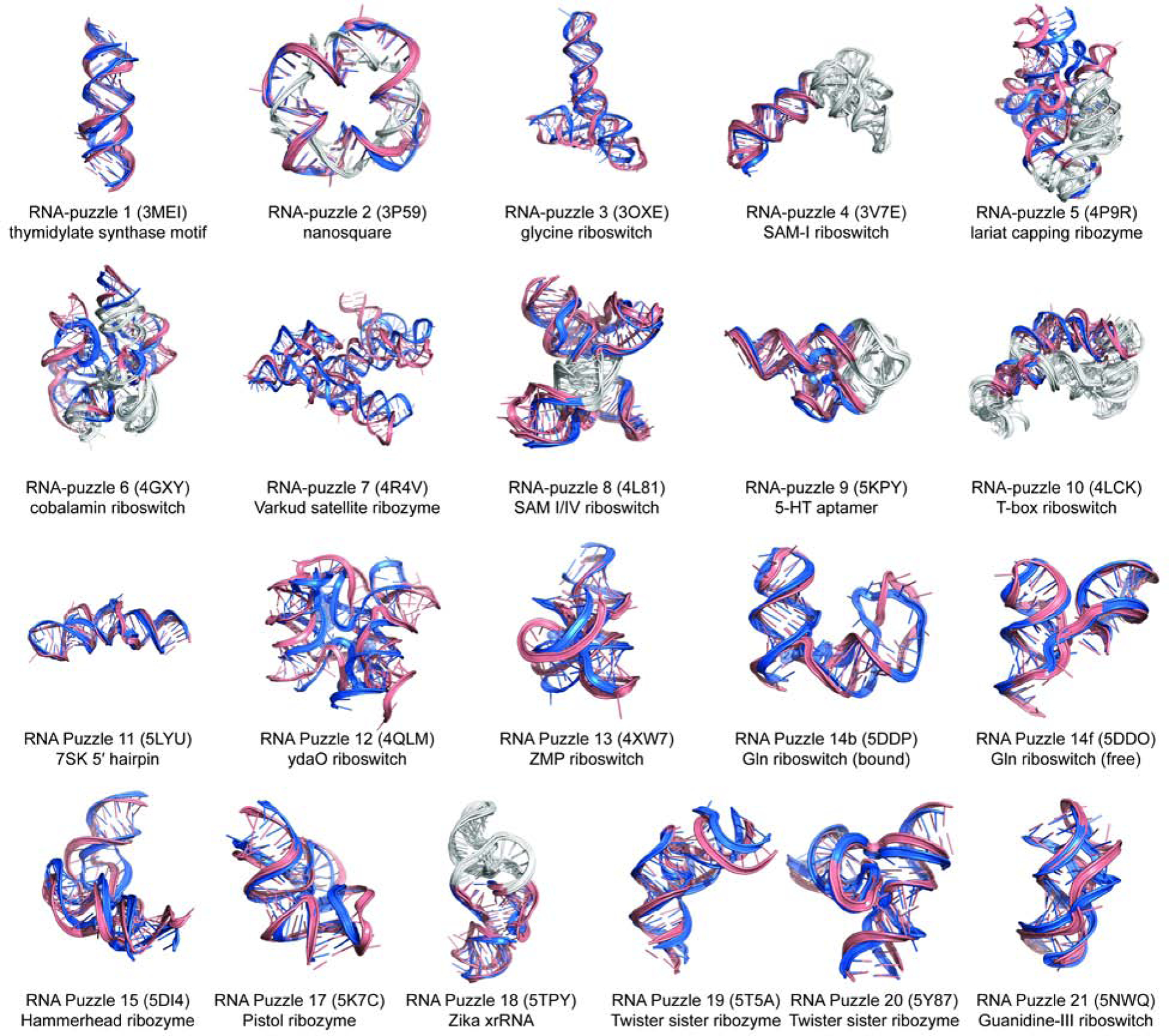
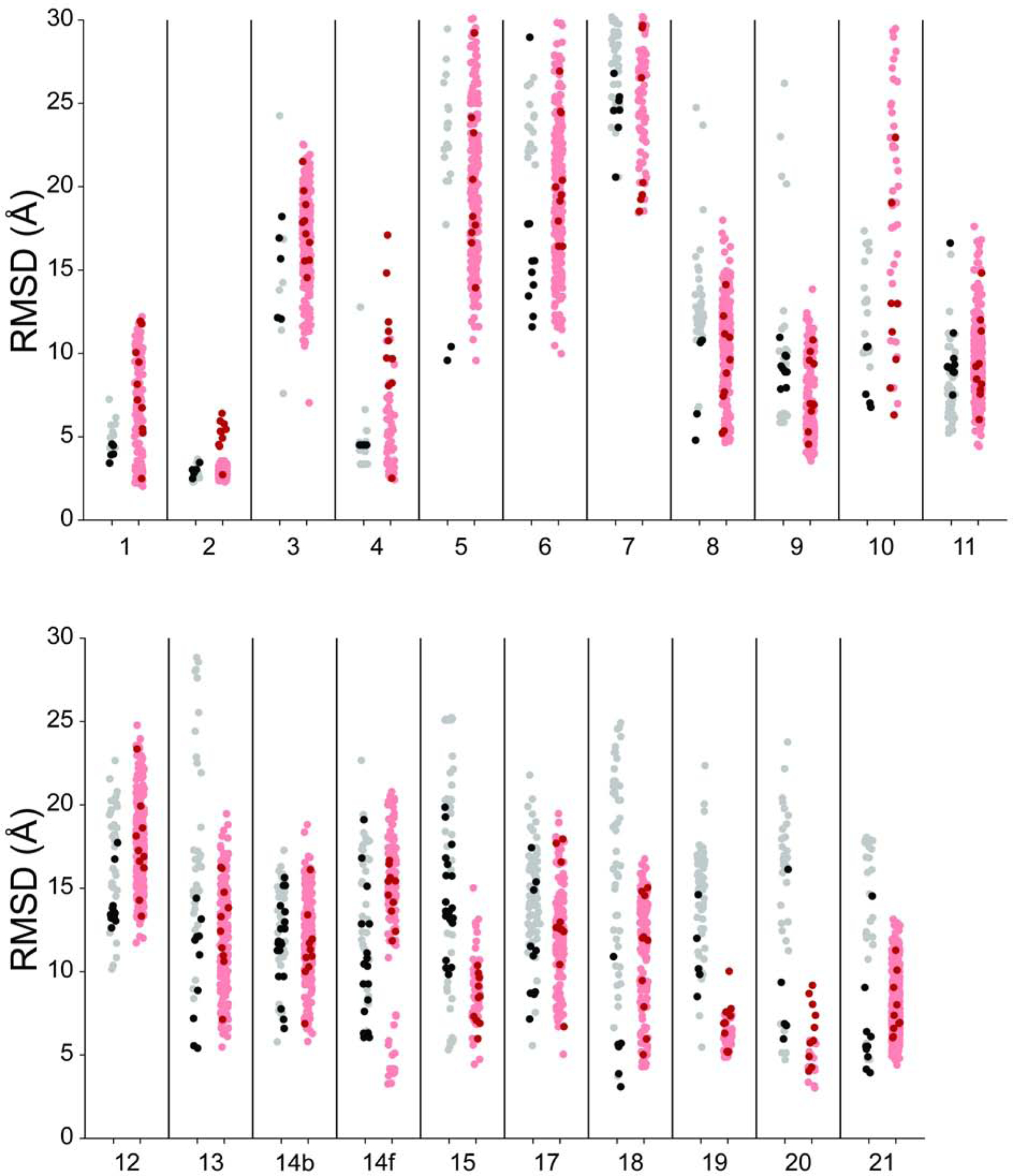
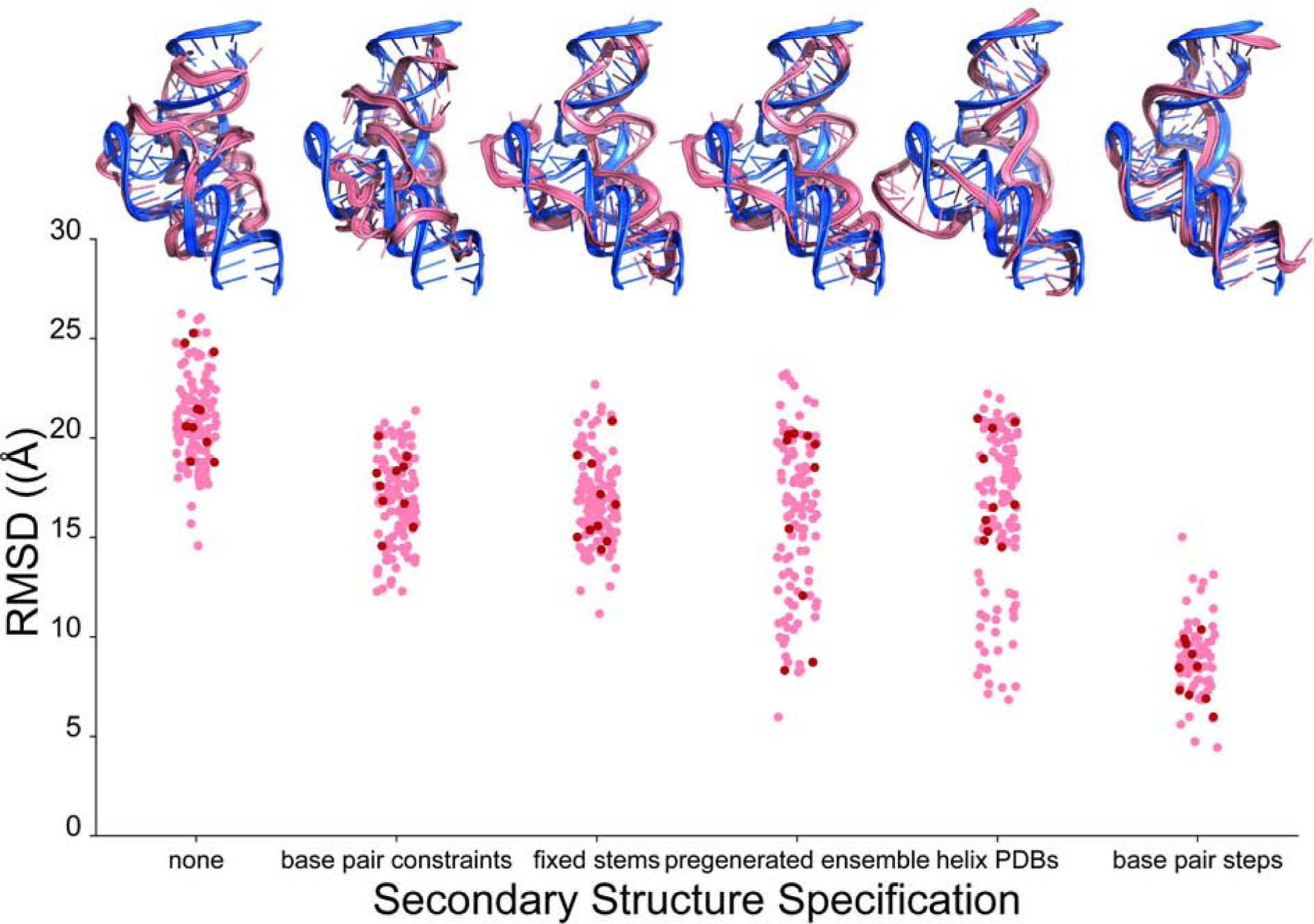
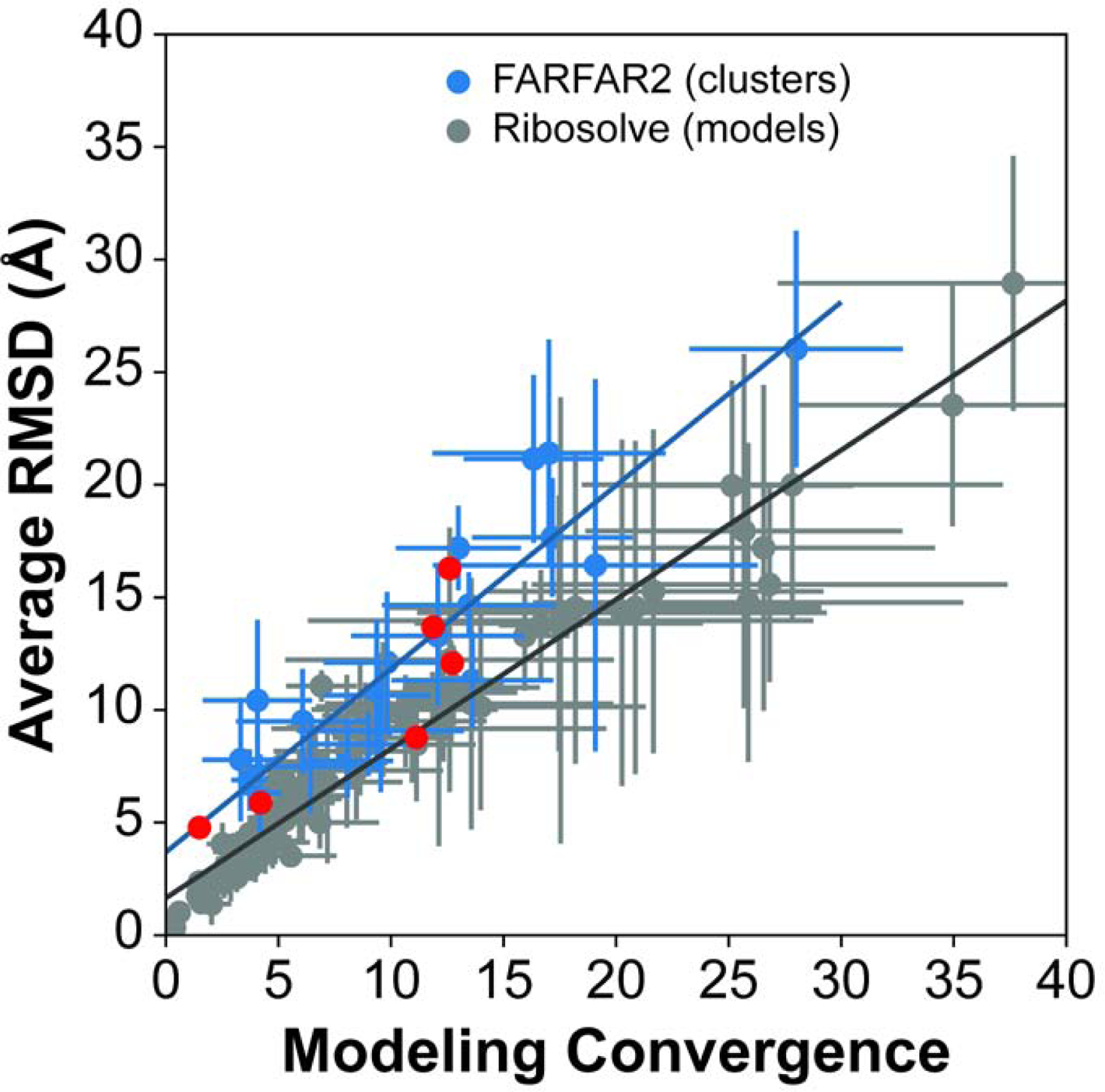
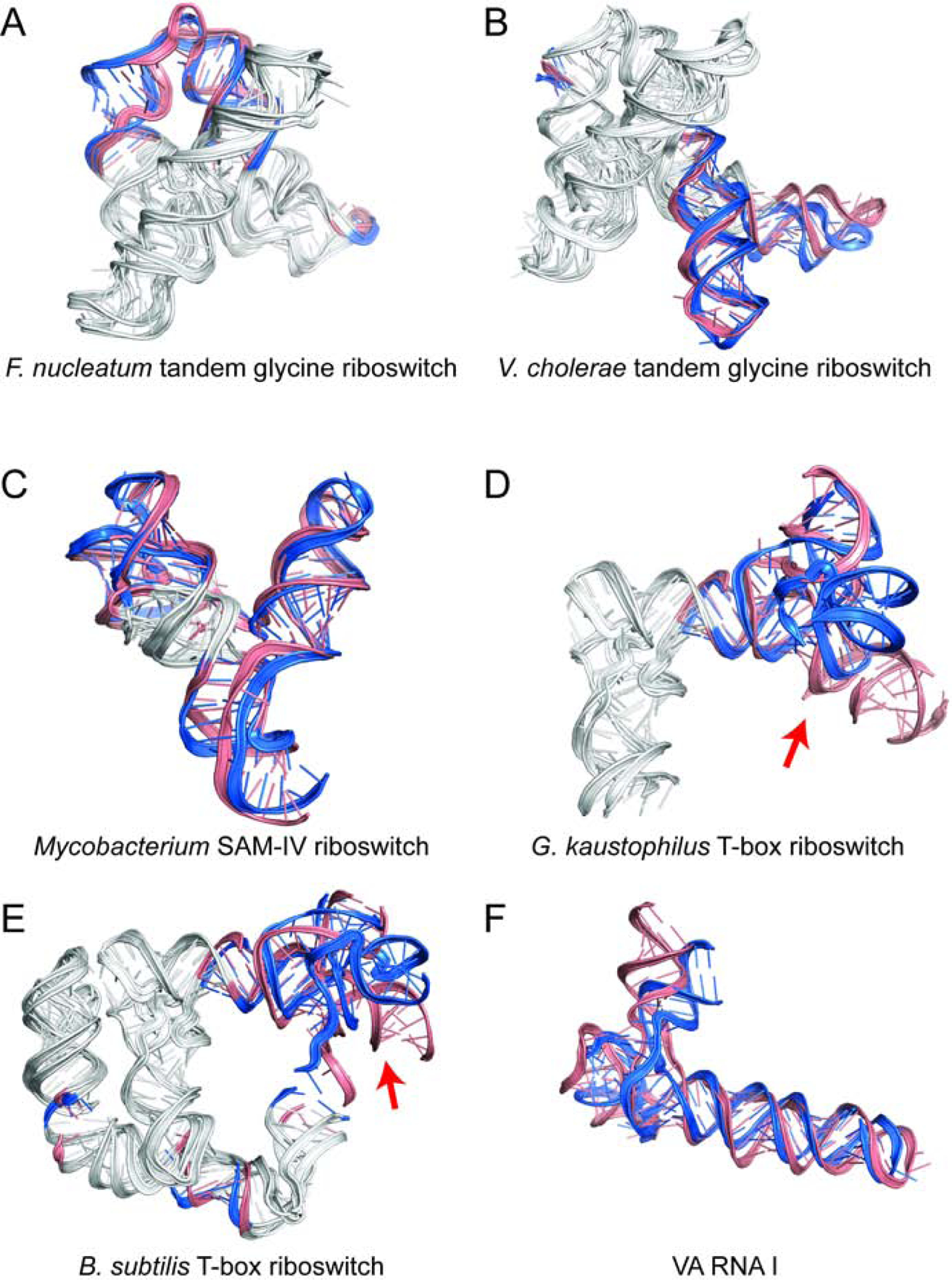
Predicting RNA three-dimensional structures from sequence could accelerate understanding of the growing number of RNA molecules being discovered across biology. Rosetta's Fragment Assembly of RNA with Full-Atom Refinement (FARFAR) has shown promise in community-wide blind RNA-Puzzle trials, but lack of a systematic and automated benchmark has left unclear what limits FARFAR performance. Here, we benchmark FARFAR2, an algorithm integrating RNA-Puzzle-inspired innovations with updated fragment libraries and helix modeling. In 16 of 21 RNA-Puzzles revisited without experimental data or expert intervention, FARFAR2 recovers native-like structures more accurate than models submitted during the RNA-Puzzles trials. Remaining bottlenecks include conformational sampling for >80-nucleotide problems and scoring function limitations more generally. Supporting these conclusions, preregistered blind models for adenovirus VA-I RNA and five riboswitch complexes predicted native-like folds with 3- to 14 Å root-mean-square deviation accuracies. We present a FARFAR2 webserver and three large model archives (FARFAR2-Classics, FARFAR2-Motifs, and FARFAR2-Puzzles) to guide future applications and advances.
Keywords: RNA; blind prediction; fragment assembly; homology modeling; structure prediction.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
Similar articles
-
RNA 3D Modeling with FARFAR2, Online.Methods Mol Biol. 2023;2586:233-249. doi: 10.1007/978-1-0716-2768-6_14. Methods Mol Biol. 2023. PMID: 36705908
-
P-FARFAR2: A multithreaded greedy approach to sampling low-energy RNA structures in Rosetta FARFAR2.Comput Biol Chem. 2023 Jun;104:107878. doi: 10.1016/j.compbiolchem.2023.107878. Epub 2023 May 3. Comput Biol Chem. 2023. PMID: 37167861
-
Modeling complex RNA tertiary folds with Rosetta.Methods Enzymol. 2015;553:35-64. doi: 10.1016/bs.mie.2014.10.051. Epub 2015 Feb 12. Methods Enzymol. 2015. PMID: 25726460
-
Energy-based RNA consensus secondary structure prediction in multiple sequence alignments.Methods Mol Biol. 2014;1097:125-41. doi: 10.1007/978-1-62703-709-9_7. Methods Mol Biol. 2014. PMID: 24639158 Review.
-
RNA Structure: Advances and Assessment of 3D Structure Prediction.Annu Rev Biophys. 2017 May 22;46:483-503. doi: 10.1146/annurev-biophys-070816-034125. Epub 2017 Mar 30. Annu Rev Biophys. 2017. PMID: 28375730 Review.
Cited by
-
Advances and opportunities in RNA structure experimental determination and computational modeling.Nat Methods. 2022 Oct;19(10):1193-1207. doi: 10.1038/s41592-022-01623-y. Epub 2022 Oct 6. Nat Methods. 2022. PMID: 36203019 Review.
-
Identification of RNA structures and their roles in RNA functions.Nat Rev Mol Cell Biol. 2024 Oct;25(10):784-801. doi: 10.1038/s41580-024-00748-6. Epub 2024 Jun 26. Nat Rev Mol Cell Biol. 2024. PMID: 38926530 Review.
-
Hierarchical Assembly of Single-Stranded RNA.J Chem Theory Comput. 2024 Mar 12;20(5):2246-2260. doi: 10.1021/acs.jctc.3c01049. Epub 2024 Feb 15. J Chem Theory Comput. 2024. PMID: 38361440 Free PMC article.
-
Conformational heterogeneity of UCAAUC RNA oligonucleotide from molecular dynamics simulations, SAXS, and NMR experiments.RNA. 2022 Jul;28(7):937-946. doi: 10.1261/rna.078888.121. Epub 2022 Apr 28. RNA. 2022. PMID: 35483823 Free PMC article.
-
Predicting RNA distance-based contact maps by integrated deep learning on physics-inferred secondary structure and evolutionary-derived mutational coupling.Bioinformatics. 2022 Aug 10;38(16):3900-3910. doi: 10.1093/bioinformatics/btac421. Bioinformatics. 2022. PMID: 35751593 Free PMC article.
References
-
- Amarasinghe GK, De Guzman RN, Turner RB, Chancellor KJ, Wu ZR, and Summers MF (2000). NMR structure of the HIV-1 nucleocapsid protein bound to stem-loop SL2 of the Ψ-RNA packaging signal. Implications for genome recognition. J. Mol. Biol 301, 491–511. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
