

Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning
- PMID: 32533916
- PMCID: PMC7384975
- DOI: 10.1016/j.cell.2020.05.037
Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning
Abstract
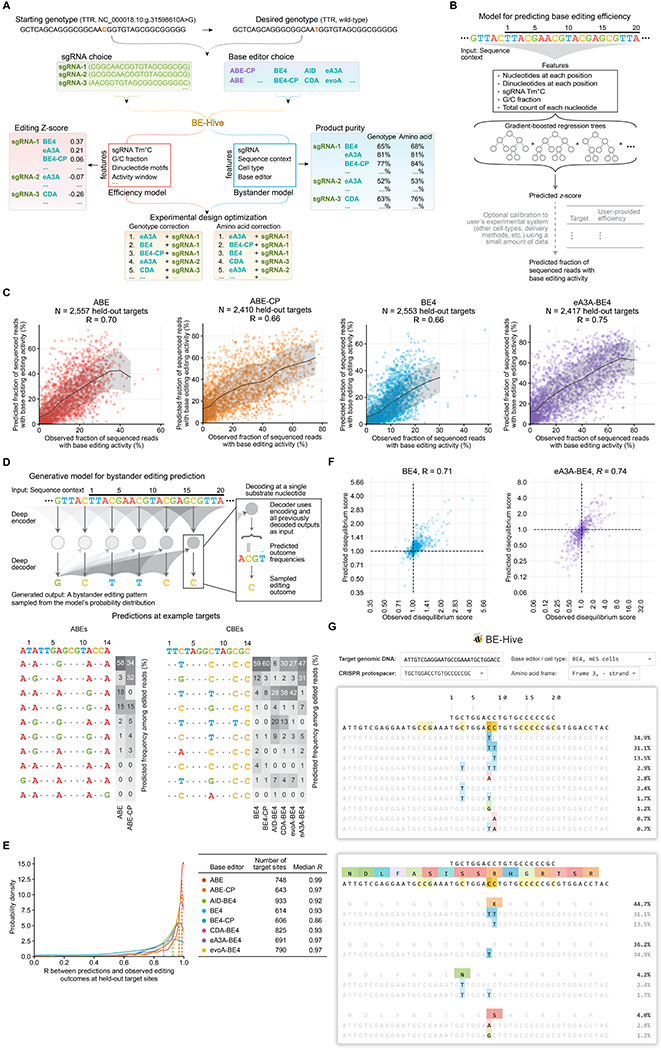
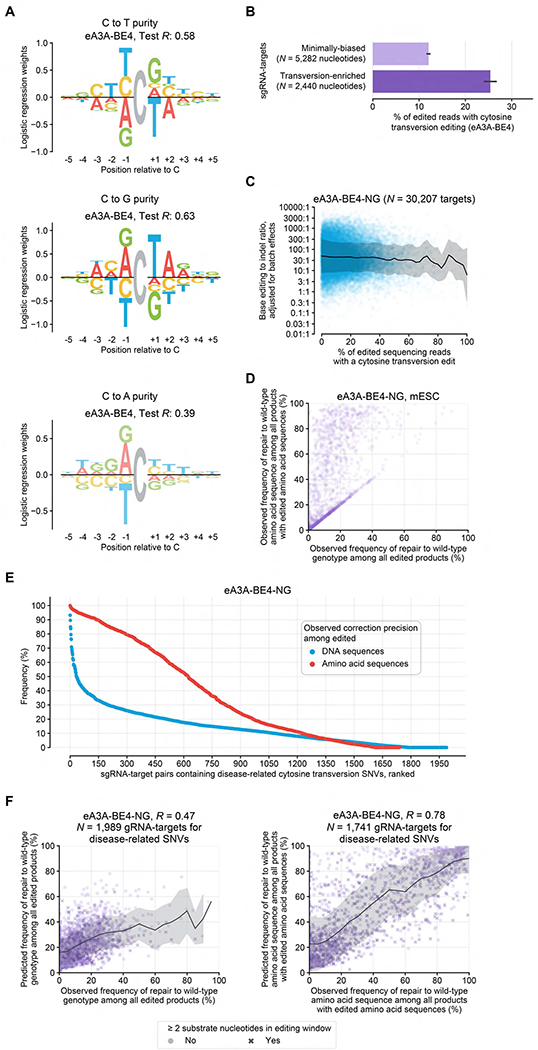
Although base editors are widely used to install targeted point mutations, the factors that determine base editing outcomes are not well understood. We characterized sequence-activity relationships of 11 cytosine and adenine base editors (CBEs and ABEs) on 38,538 genomically integrated targets in mammalian cells and used the resulting outcomes to train BE-Hive, a machine learning model that accurately predicts base editing genotypic outcomes (R ≈ 0.9) and efficiency (R ≈ 0.7). We corrected 3,388 disease-associated SNVs with ≥90% precision, including 675 alleles with bystander nucleotides that BE-Hive correctly predicted would not be edited. We discovered determinants of previously unpredictable C-to-G, or C-to-A editing and used these discoveries to correct coding sequences of 174 pathogenic transversion SNVs with ≥90% precision. Finally, we used insights from BE-Hive to engineer novel CBE variants that modulate editing outcomes. These discoveries illuminate base editing, enable editing at previously intractable targets, and provide new base editors with improved editing capabilities.
Keywords: base editing; disease correction; machine learning; precision genome editing; transversion base editing.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests D.R.L. is a consultant and co-founder of Beam Therapeutics, Prime Medicine, Editas Medicine, and Pairwise Plants, companies that use genome editing technologies. The authors have filed a patent application on aspects of this work.
Figures





Comment in
-
A Navigation System for Base Editing: Are We There Yet?CRISPR J. 2020 Aug;3(4):224-225. doi: 10.1089/crispr.2020.29097.dcb. CRISPR J. 2020. PMID: 32833537 No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
