

High-Throughput Reclassification of SCN5A Variants
- PMID: 32533946
- PMCID: PMC7332654
- DOI: 10.1016/j.ajhg.2020.05.015
High-Throughput Reclassification of SCN5A Variants
Abstract
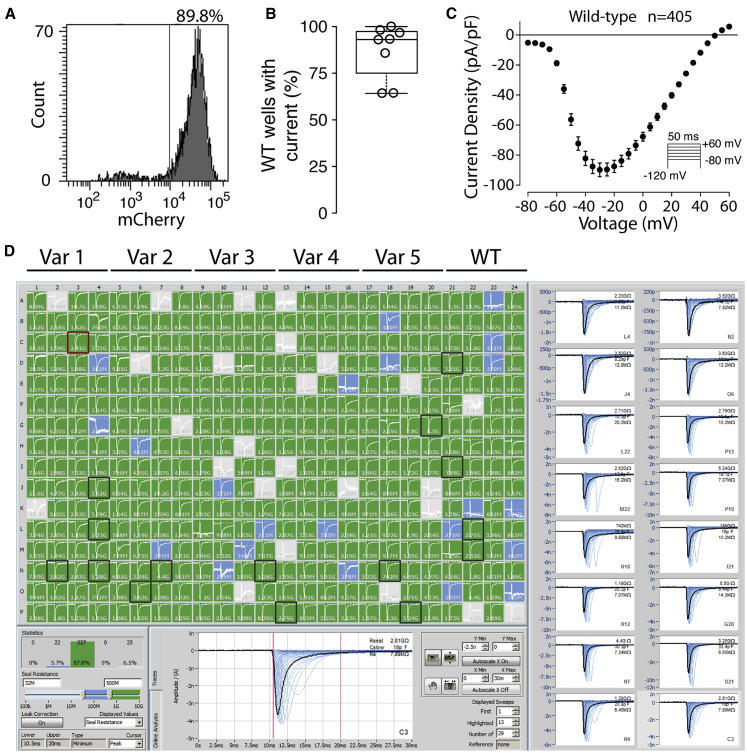
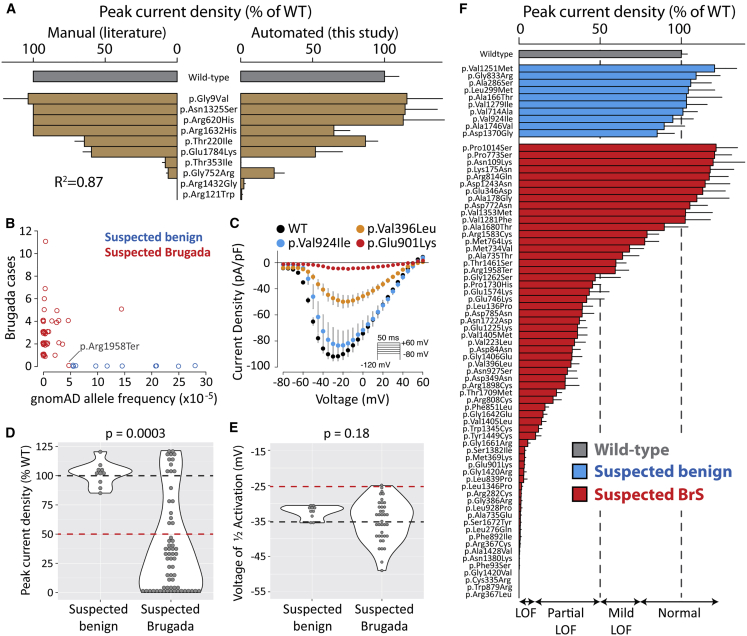
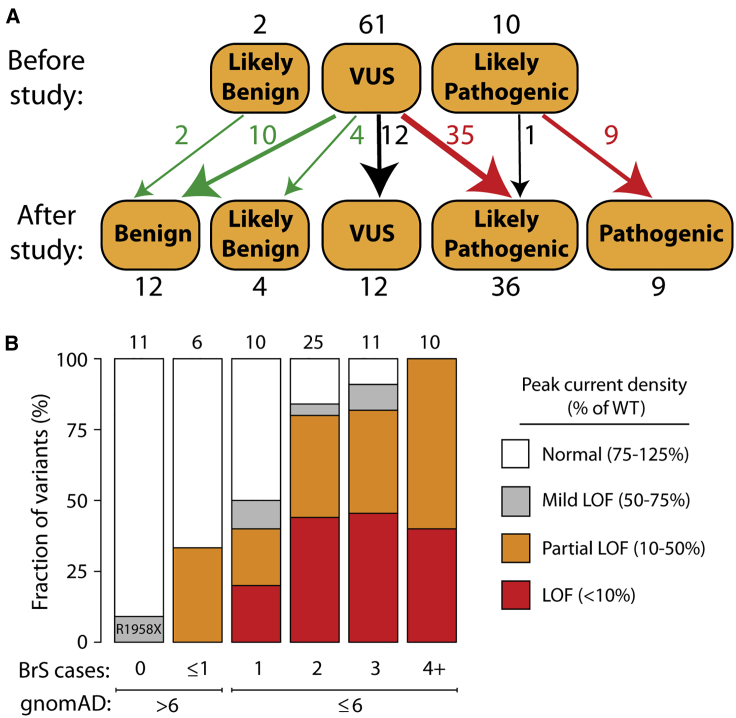
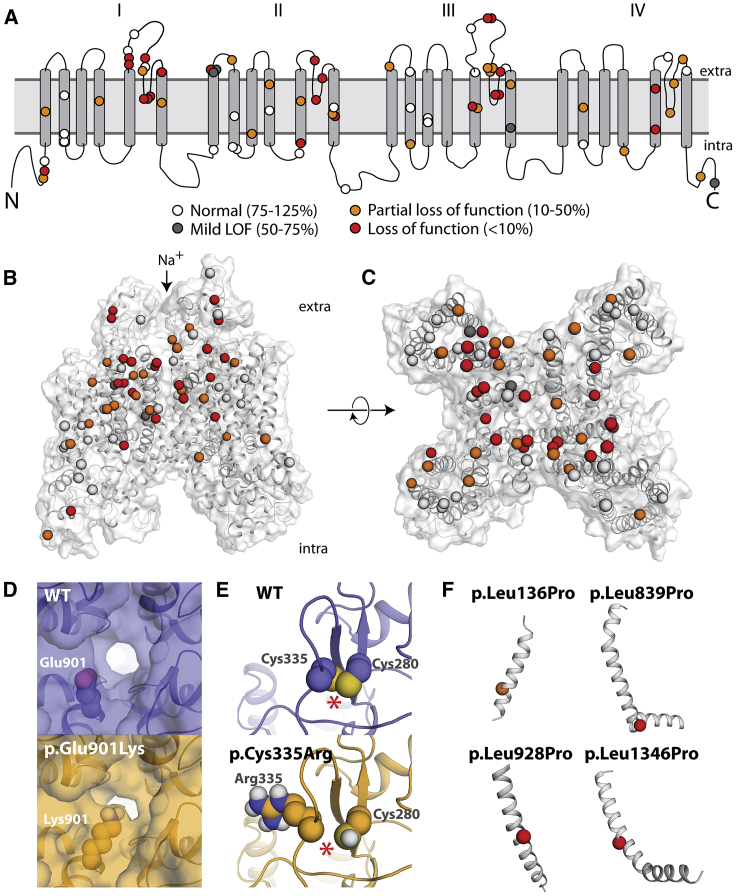
Partial or complete loss-of-function variants in SCN5A are the most common genetic cause of the arrhythmia disorder Brugada syndrome (BrS1). However, the pathogenicity of SCN5A variants is often unknown or disputed; 80% of the 1,390 SCN5A missense variants observed in at least one individual to date are variants of uncertain significance (VUSs). The designation of VUS is a barrier to the use of sequence data in clinical care. We selected 83 variants: 10 previously studied control variants, 10 suspected benign variants, and 63 suspected Brugada syndrome-associated variants, selected on the basis of their frequency in the general population and in individuals with Brugada syndrome. We used high-throughput automated patch clamping to study the function of the 83 variants, with the goal of reclassifying variants with functional data. The ten previously studied controls had functional properties concordant with published manual patch clamp data. All 10 suspected benign variants had wild-type-like function. 22 suspected BrS variants had loss of channel function (<10% normalized peak current) and 22 variants had partial loss of function (10%-50% normalized peak current). The previously unstudied variants were initially classified as likely benign (n = 2), likely pathogenic (n = 10), or VUSs (n = 61). After the patch clamp studies, 16 variants were benign/likely benign, 45 were pathogenic/likely pathogenic, and only 12 were still VUSs. Structural modeling identified likely mechanisms for loss of function including altered thermostability and disruptions to alpha helices, disulfide bonds, or the permeation pore. High-throughput patch clamping enabled reclassification of the majority of tested VUSs in SCN5A.
Keywords: Brugada syndrome; Na(V)1.5; SCN5A; high-throughput; patch clamp; variant of uncertain significance.
Copyright © 2020 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Kapplinger J.D., Tester D.J., Alders M., Benito B., Berthet M., Brugada J., Brugada P., Fressart V., Guerchicoff A., Harris-Kerr C. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7:33–46. - PMC - PubMed
-
- Kapplinger J.D., Tester D.J., Salisbury B.A., Carr J.L., Harris-Kerr C., Pollevick G.D., Wilde A.A.M., Ackerman M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm. 2009;6:1297–1303. - PMC - PubMed
-
- Bezzina C.R., Rook M.B., Groenewegen W.A., Herfst L.J., van der Wal A.C., Lam J., Jongsma H.J., Wilde A.A., Mannens M.M. Compound heterozygosity for mutations (W156X and R225W) in SCN5A associated with severe cardiac conduction disturbances and degenerative changes in the conduction system. Circ. Res. 2003;92:159–168. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
