

Hypercapnia Suppresses Macrophage Antiviral Activity and Increases Mortality of Influenza A Infection via Akt1
- PMID: 32540997
- PMCID: PMC7343622
- DOI: 10.4049/jimmunol.2000085
Hypercapnia Suppresses Macrophage Antiviral Activity and Increases Mortality of Influenza A Infection via Akt1
Abstract
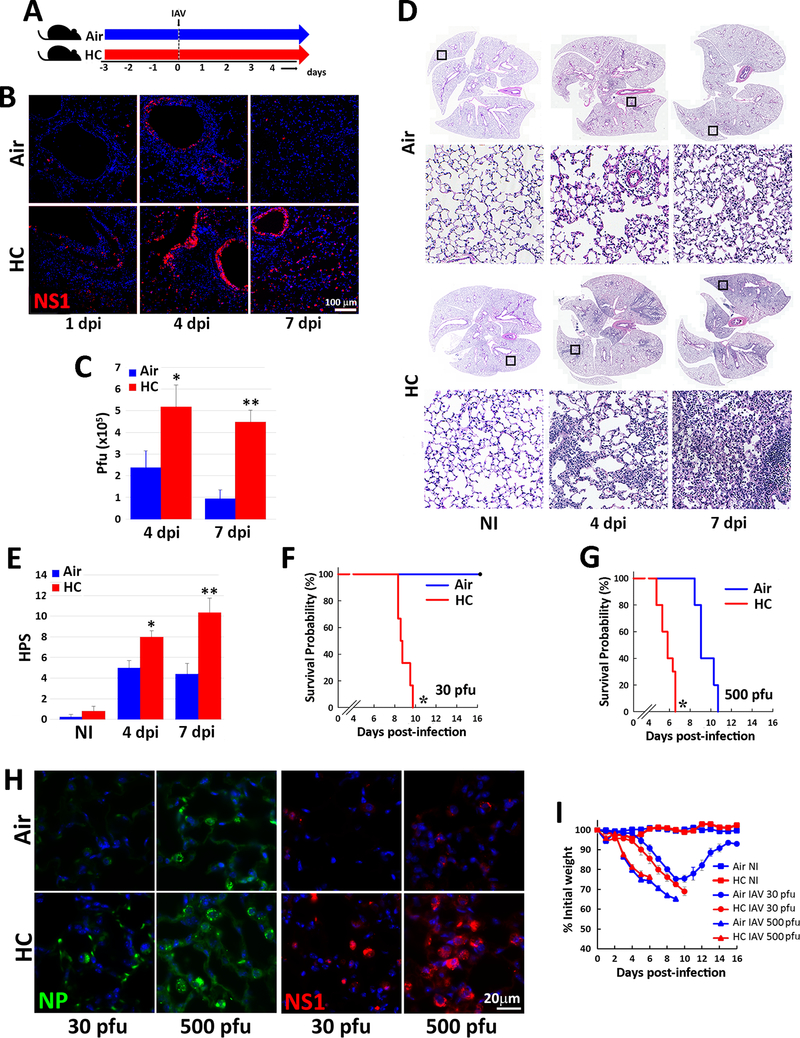
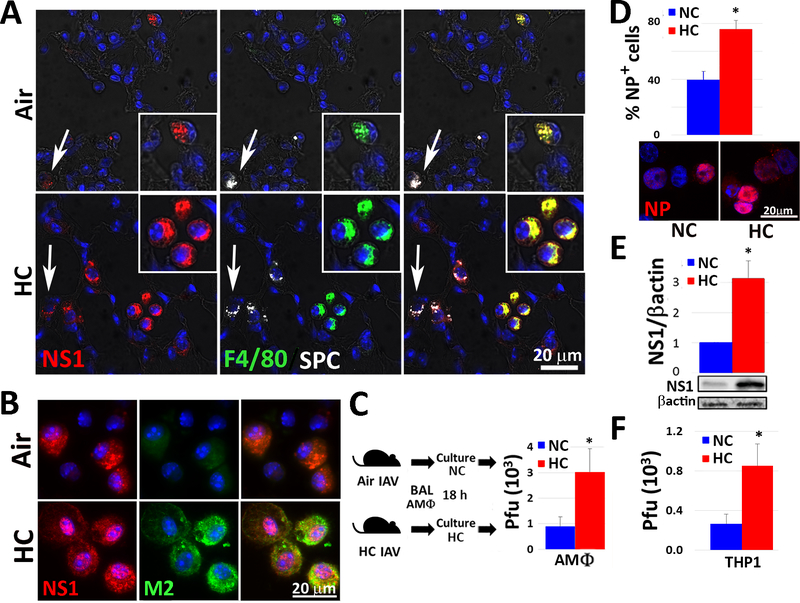
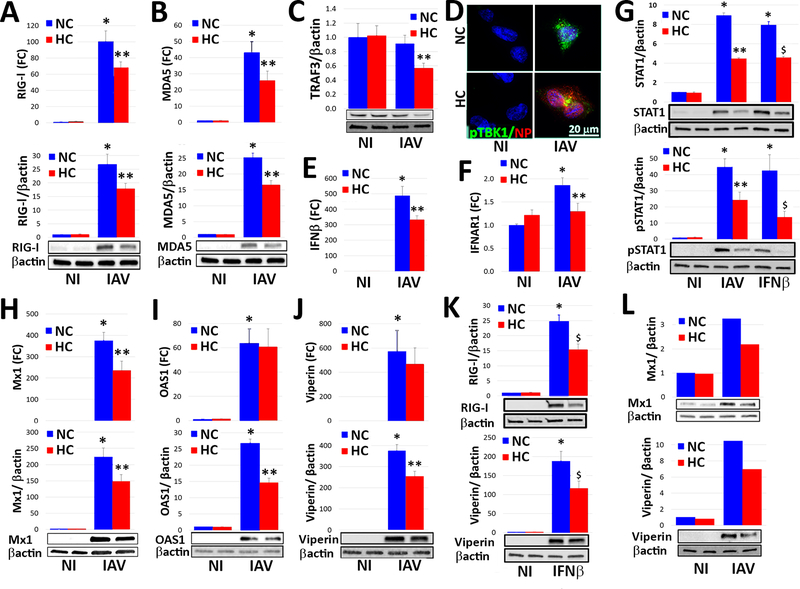
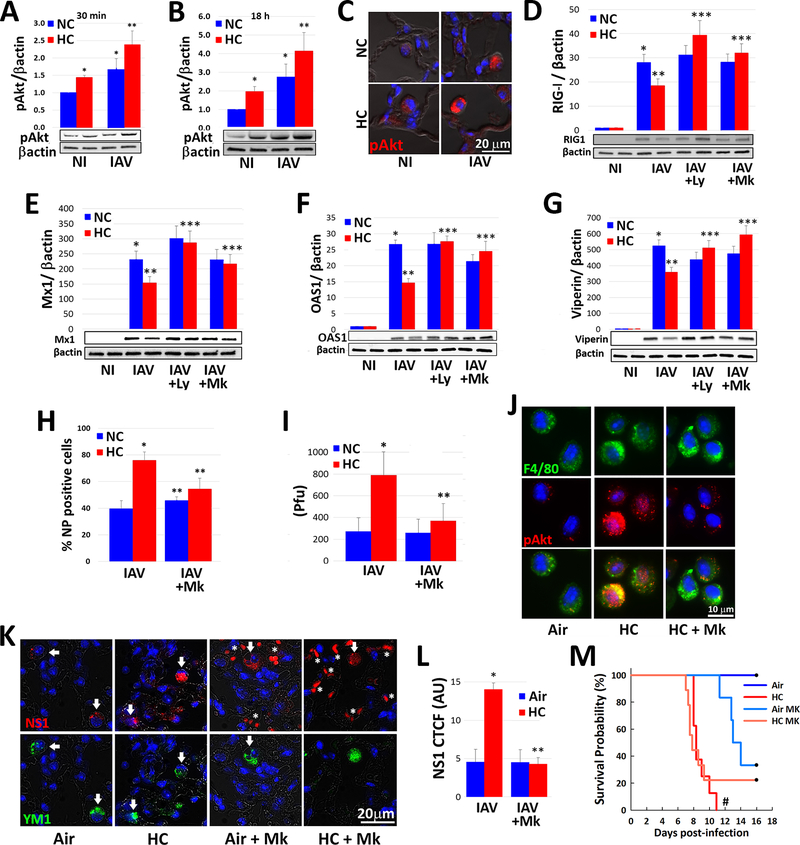
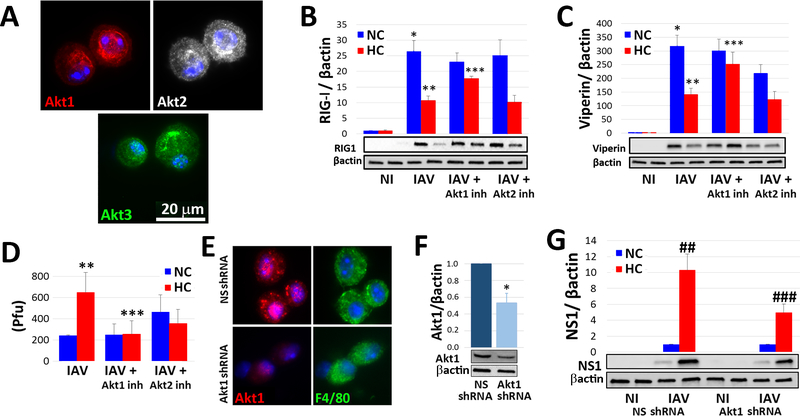
Hypercapnia (HC), elevation of the partial pressure of CO2 in blood and tissues, is a risk factor for mortality in patients with severe acute and chronic lung diseases. We previously showed that HC inhibits multiple macrophage and neutrophil antimicrobial functions and increases the mortality of bacterial pneumonia in mice. In this study, we show that normoxic HC increases viral replication, lung injury, and mortality in mice infected with influenza A virus (IAV). Elevated CO2 increased IAV replication and inhibited antiviral gene and protein expression in macrophages in vivo and in vitro. HC potentiated IAV-induced activation of Akt, whereas specific pharmacologic inhibition or short hairpin RNA knockdown of Akt1 in alveolar macrophages blocked HC's effects on IAV growth and the macrophage antiviral response. Our findings suggest that targeting Akt1 or the downstream pathways through which elevated CO2 signals could enhance macrophage antiviral host defense and improve clinical outcomes in hypercapnic patients with advanced lung disease.
Copyright © 2020 by The American Association of Immunologists, Inc.
Figures
References
-
- Heron M 2019. Deaths: Leading Causes for 2017. National Vital Statistics Reports 68: 1–74. - PubMed
-
- 2018. The top 10 causes of death. World Health Organization, https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death.
-
- Moser KM, Shibel EM, and Beamon AJ 1973. Acute respiratory failure in obstructive lung disease. Long-term survival after treatment in an intensive care unit. JAMA 225: 705–707. - PubMed
-
- Martin TR, Lewis SW, and Albert RK 1982. The prognosis of patients with chronic obstructive pulmonary disease after hospitalization for acute respiratory failure. Chest 82: 310–314. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
