

Recurrent inversion toggling and great ape genome evolution
- PMID: 32541924
- PMCID: PMC7415573
- DOI: 10.1038/s41588-020-0646-x
Recurrent inversion toggling and great ape genome evolution
Abstract
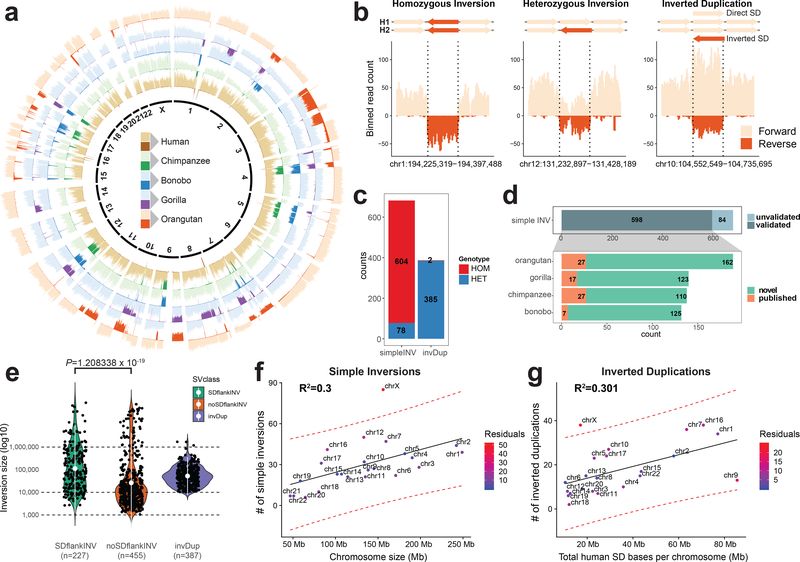
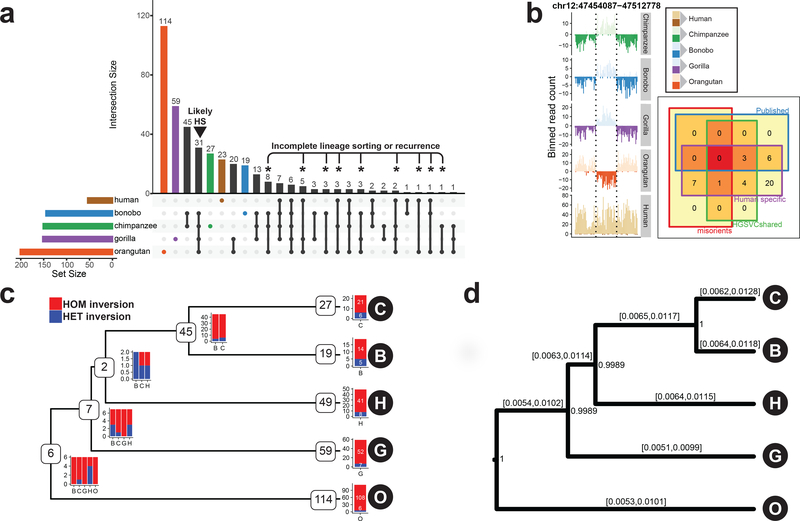
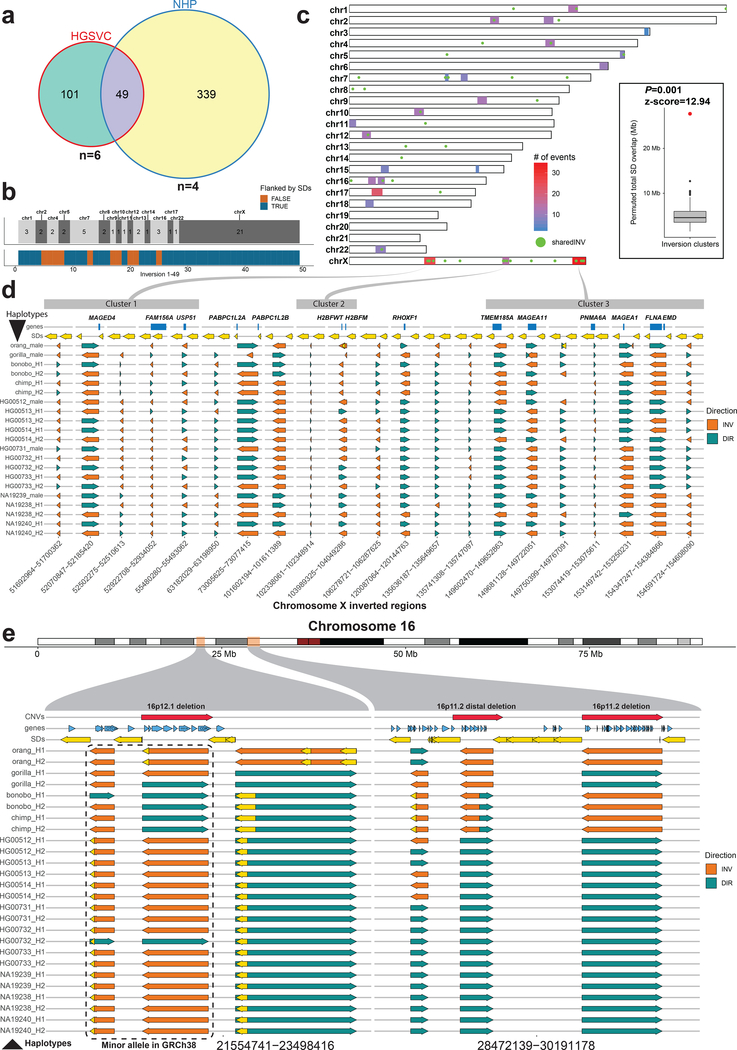
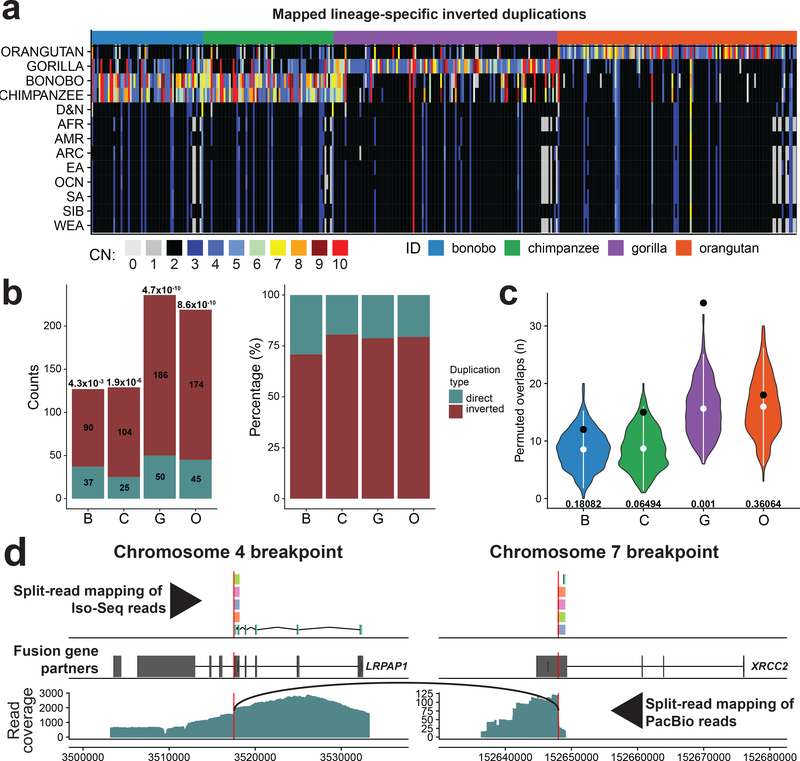
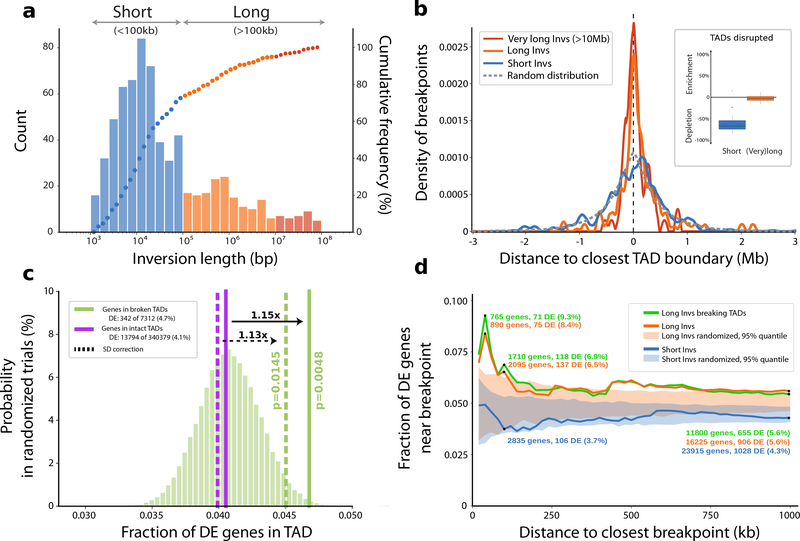
Inversions play an important role in disease and evolution but are difficult to characterize because their breakpoints map to large repeats. We increased by sixfold the number (n = 1,069) of previously reported great ape inversions by using single-cell DNA template strand and long-read sequencing. We find that the X chromosome is most enriched (2.5-fold) for inversions, on the basis of its size and duplication content. There is an excess of differentially expressed primate genes near the breakpoints of large (>100 kilobases (kb)) inversions but not smaller events. We show that when great ape lineage-specific duplications emerge, they preferentially (approximately 75%) occur in an inverted orientation compared to that at their ancestral locus. We construct megabase-pair scale haplotypes for individual chromosomes and identify 23 genomic regions that have recurrently toggled between a direct and an inverted state over 15 million years. The direct orientation is most frequently the derived state for human polymorphisms that predispose to recurrent copy number variants associated with neurodevelopmental disease.
Conflict of interest statement
COMPETING INTERESTS
E.E.E. is on the scientific advisory board (SAB) of DNAnexus, Inc.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
