

A robust benchmark for detection of germline large deletions and insertions
- PMID: 32541955
- PMCID: PMC8454654
- DOI: 10.1038/s41587-020-0538-8
A robust benchmark for detection of germline large deletions and insertions
Erratum in
-
Author Correction: A robust benchmark for detection of germline large deletions and insertions.Nat Biotechnol. 2020 Nov;38(11):1357. doi: 10.1038/s41587-020-0640-y. Nat Biotechnol. 2020. PMID: 32699374
Abstract
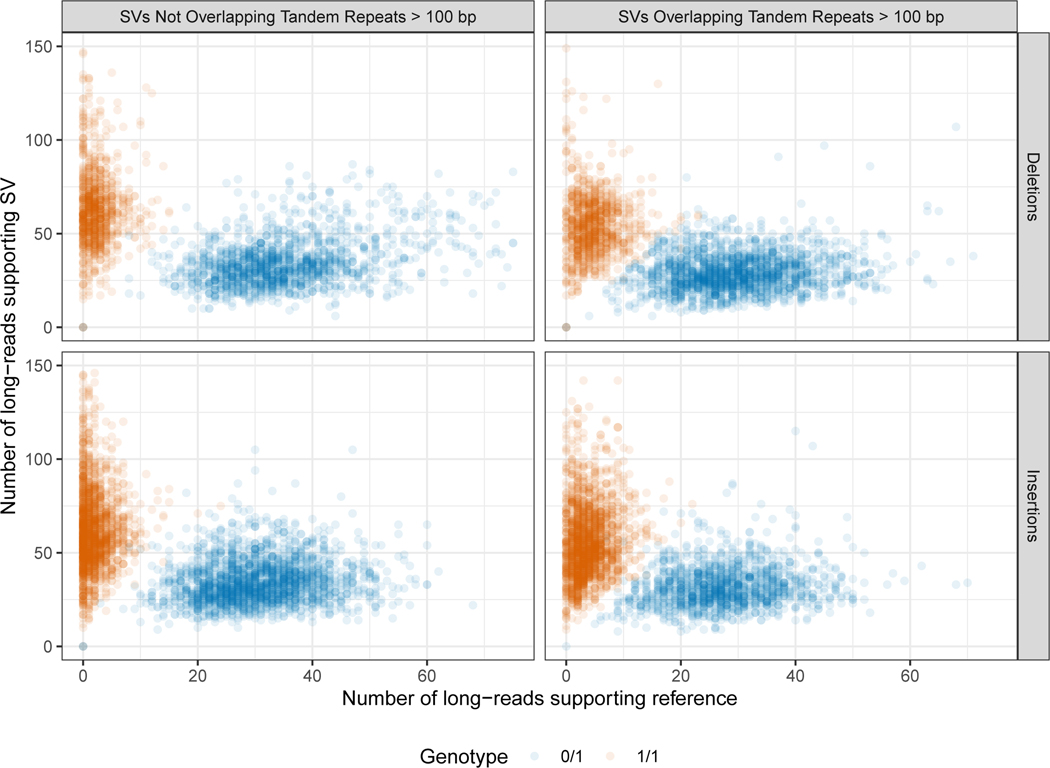
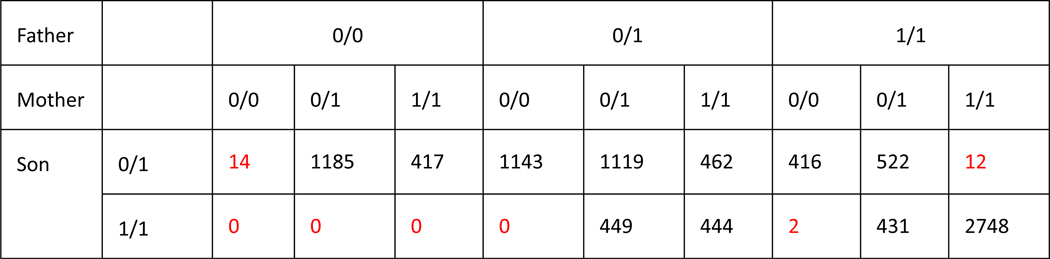
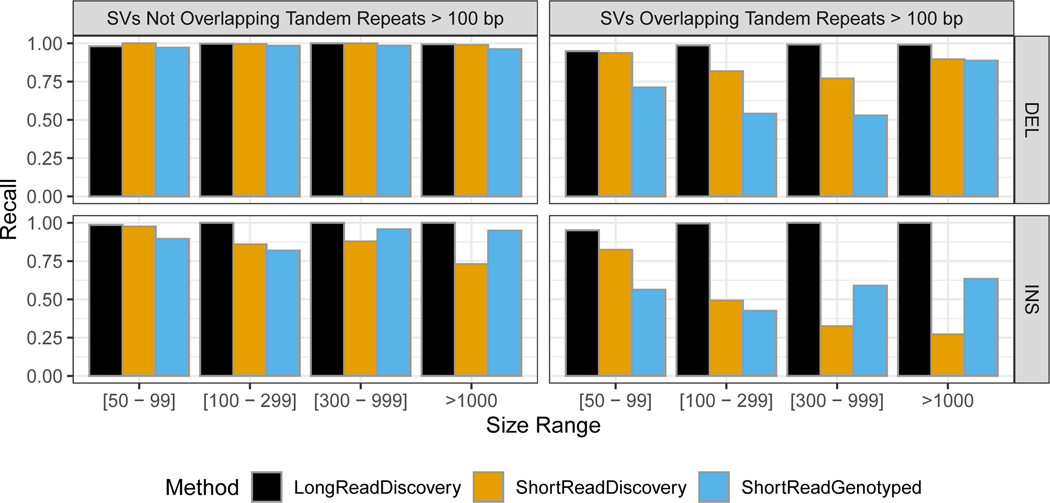
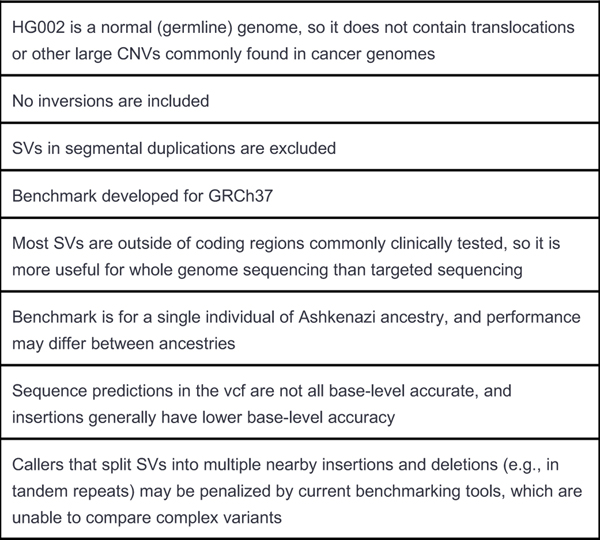
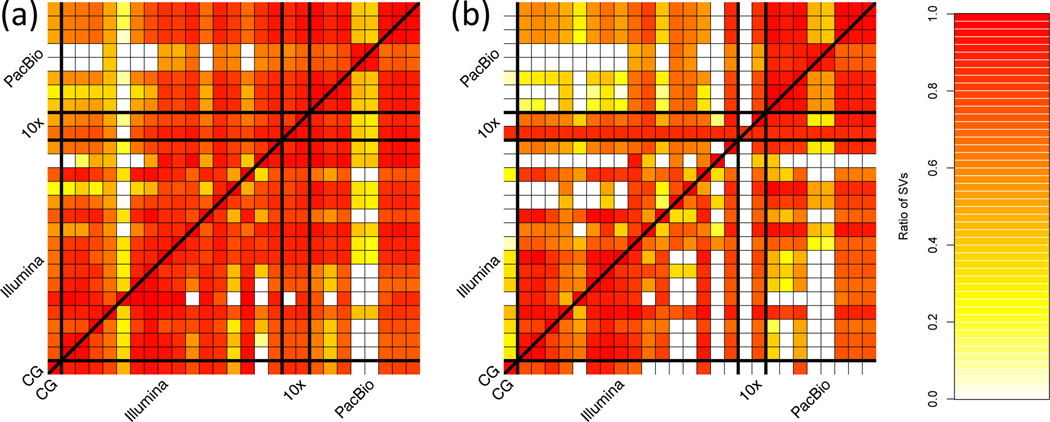
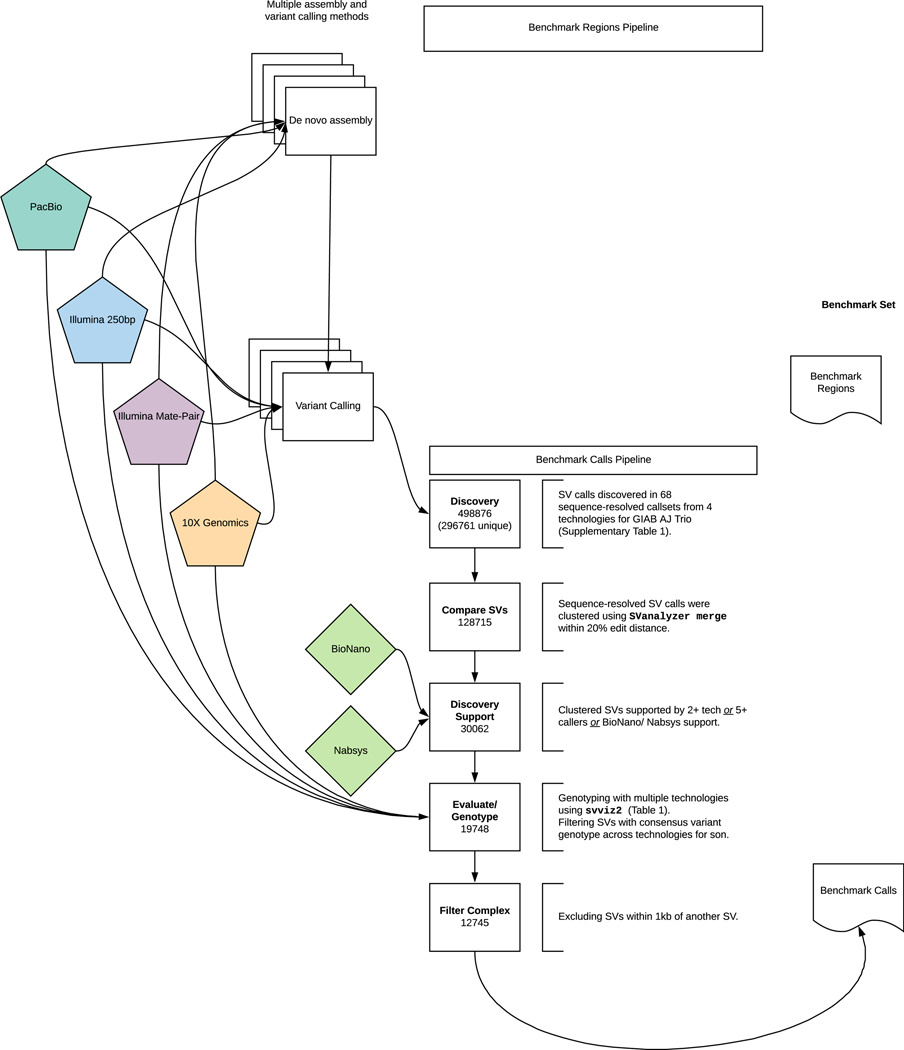
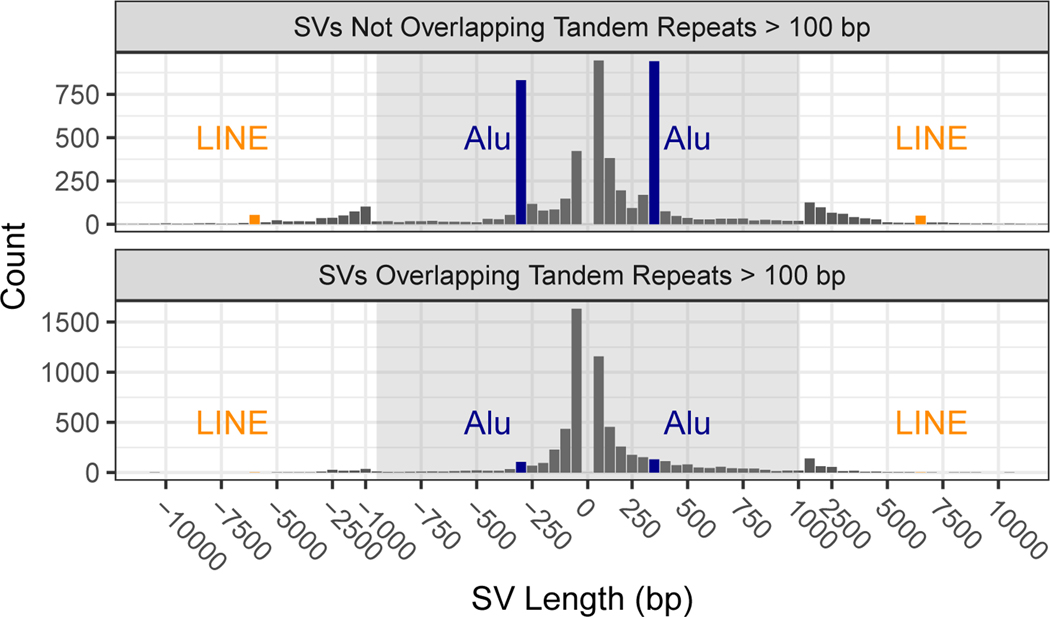
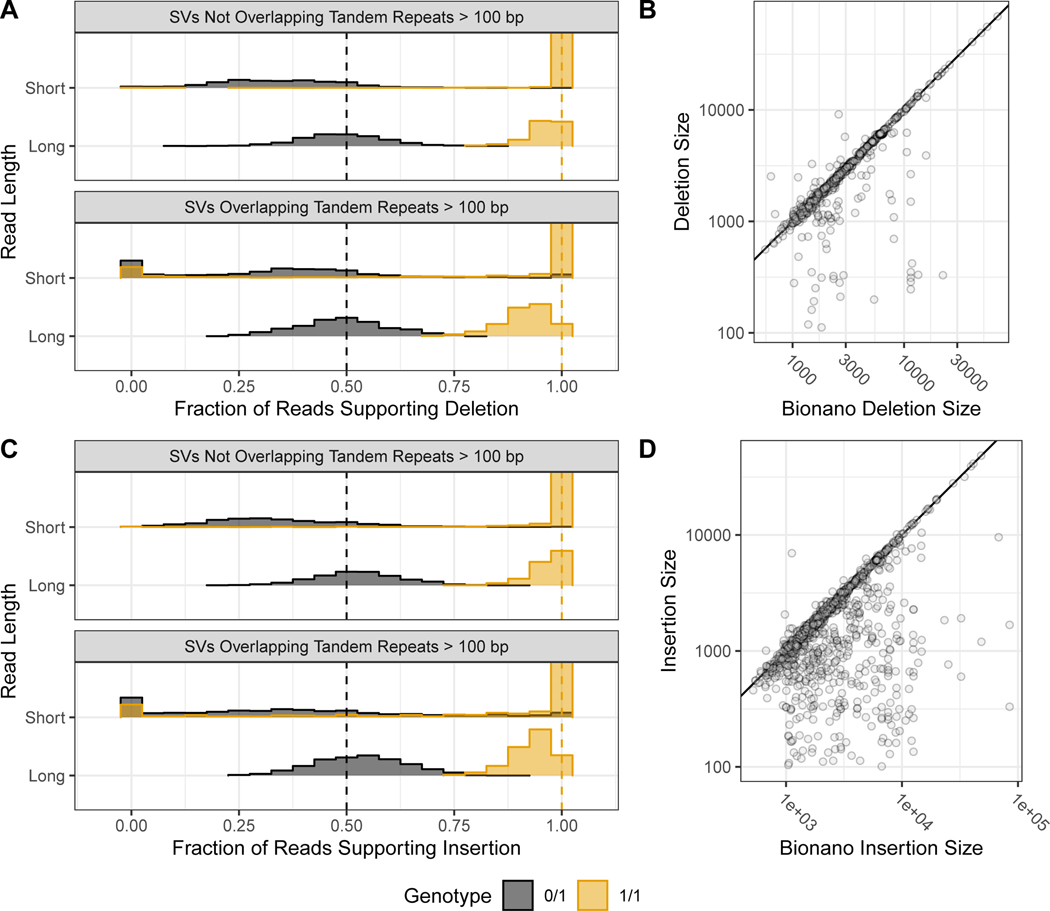
New technologies and analysis methods are enabling genomic structural variants (SVs) to be detected with ever-increasing accuracy, resolution and comprehensiveness. To help translate these methods to routine research and clinical practice, we developed a sequence-resolved benchmark set for identification of both false-negative and false-positive germline large insertions and deletions. To create this benchmark for a broadly consented son in a Personal Genome Project trio with broadly available cells and DNA, the Genome in a Bottle Consortium integrated 19 sequence-resolved variant calling methods from diverse technologies. The final benchmark set contains 12,745 isolated, sequence-resolved insertion (7,281) and deletion (5,464) calls ≥50 base pairs (bp). The Tier 1 benchmark regions, for which any extra calls are putative false positives, cover 2.51 Gbp and 5,262 insertions and 4,095 deletions supported by ≥1 diploid assembly. We demonstrate that the benchmark set reliably identifies false negatives and false positives in high-quality SV callsets from short-, linked- and long-read sequencing and optical mapping.
Figures
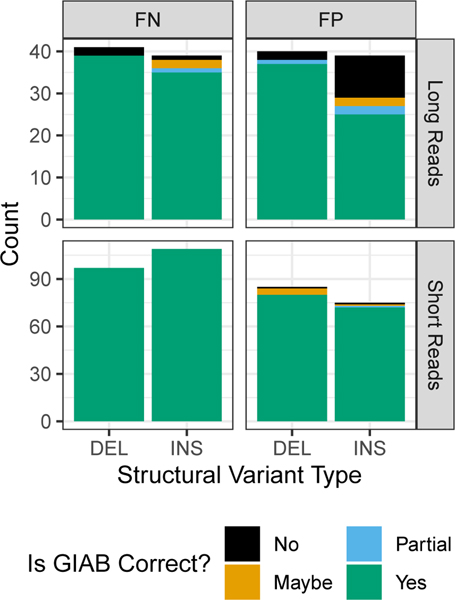
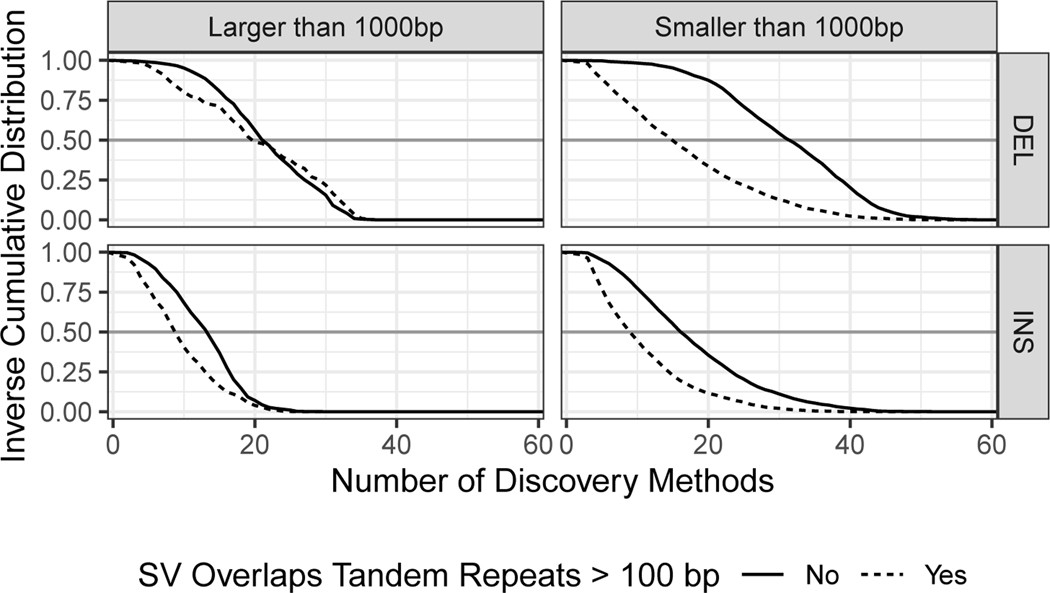
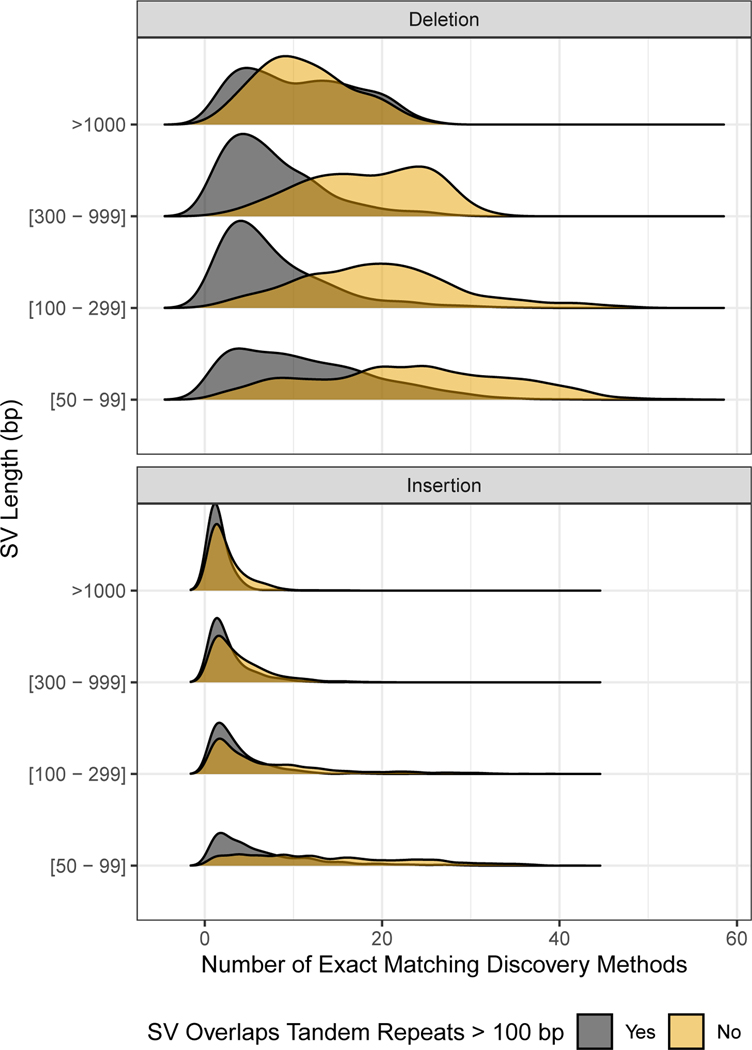
References
-
- Roses AD et al.Structural variants can be more informative for disease diagnostics, prognostics and translation than current SNP mapping and exon sequencing. Expert Opin. Drug Metab. Toxicol 12, 135–147 (2016). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
