

CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity
- PMID: 32541958
- PMCID: PMC7652380
- DOI: 10.1038/s41587-020-0555-7
CHANGE-seq reveals genetic and epigenetic effects on CRISPR-Cas9 genome-wide activity
Abstract
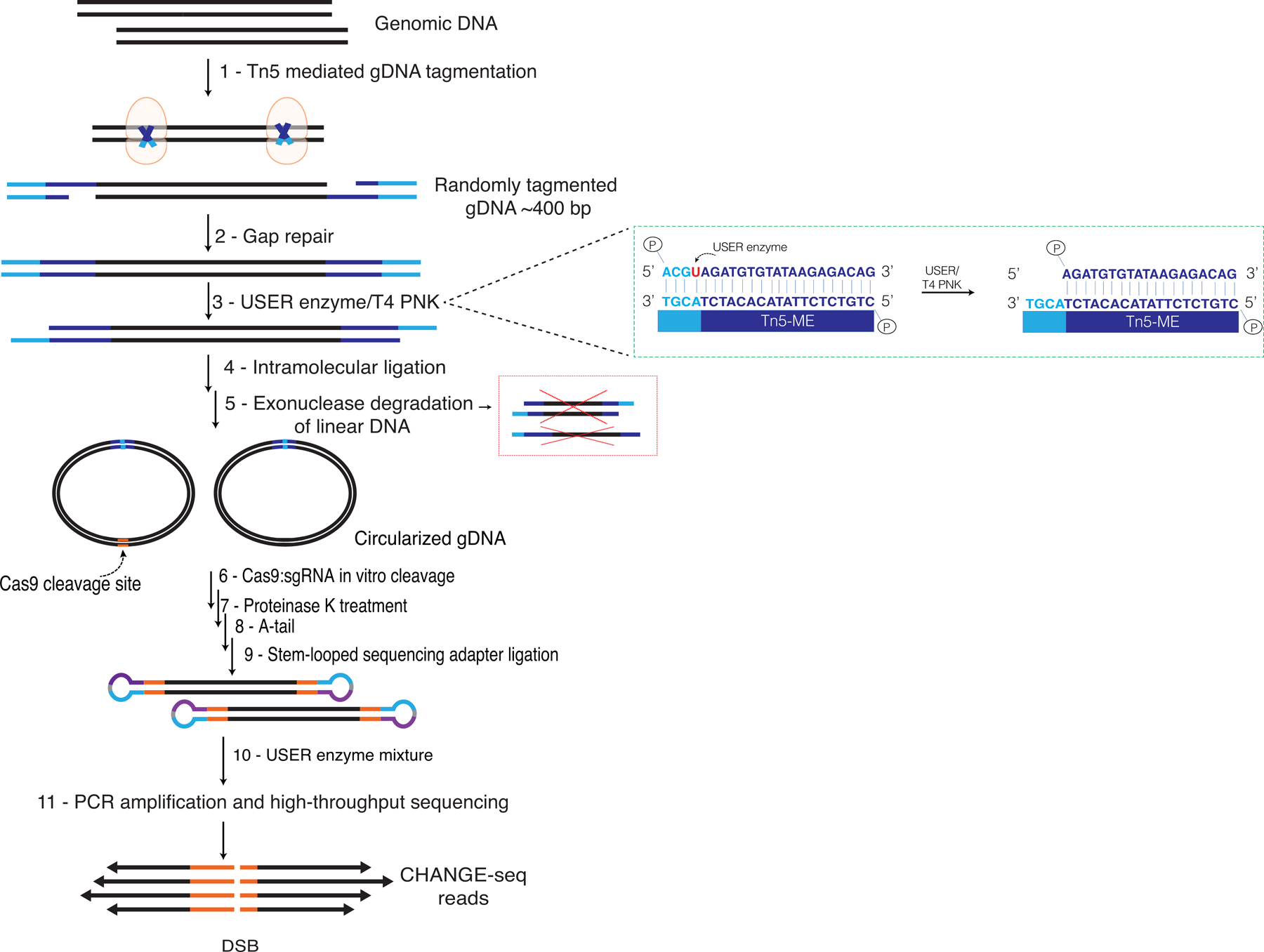
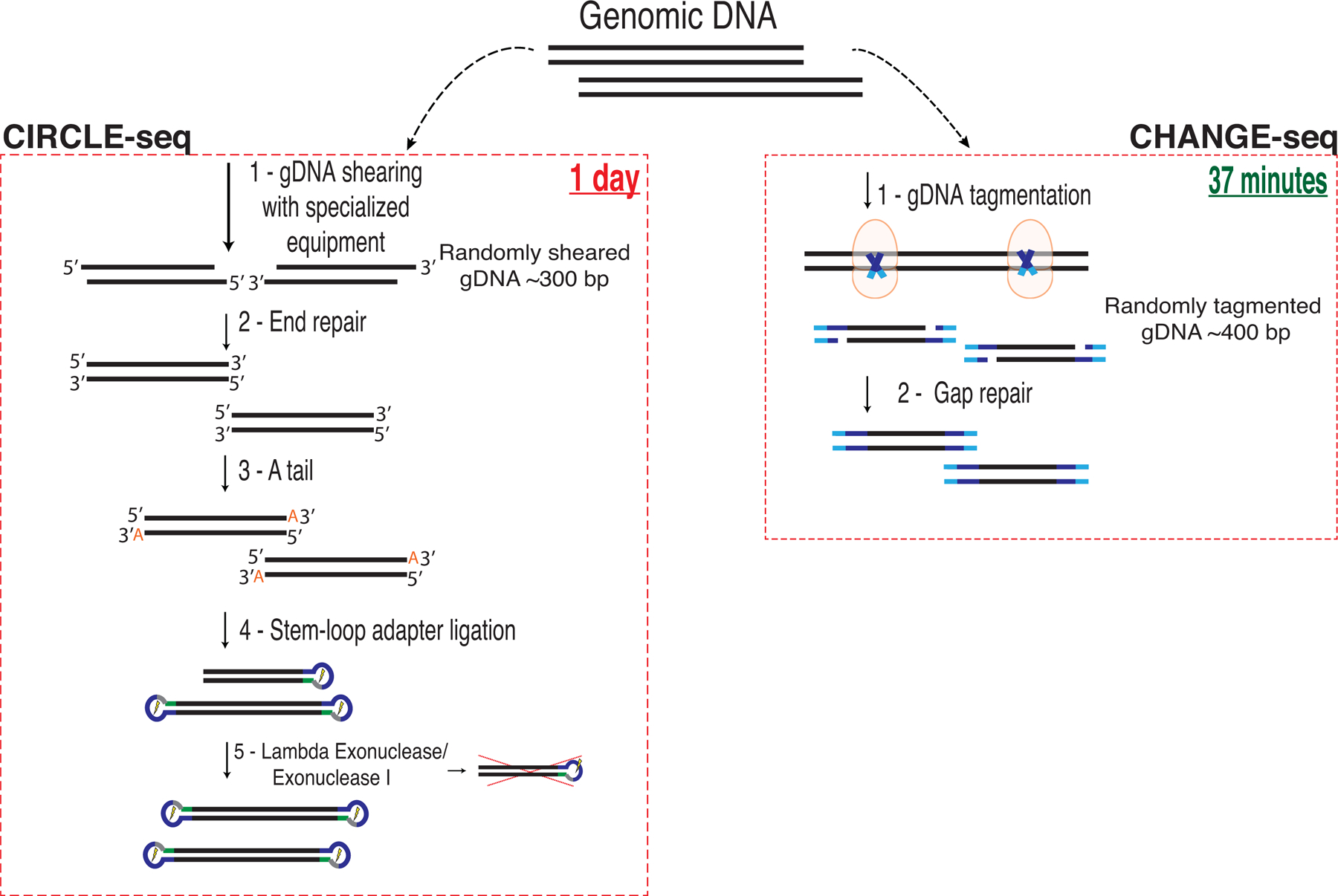
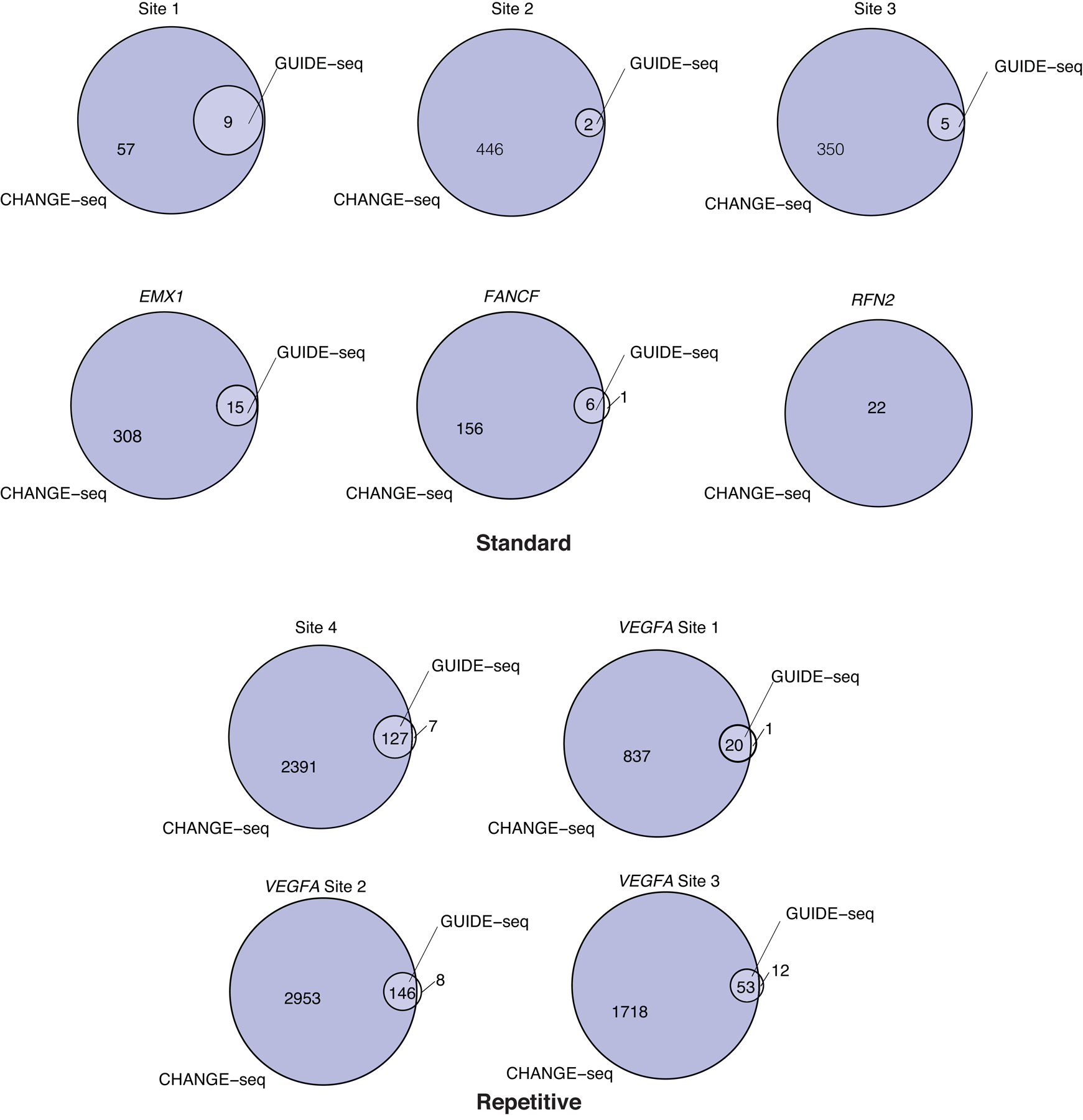
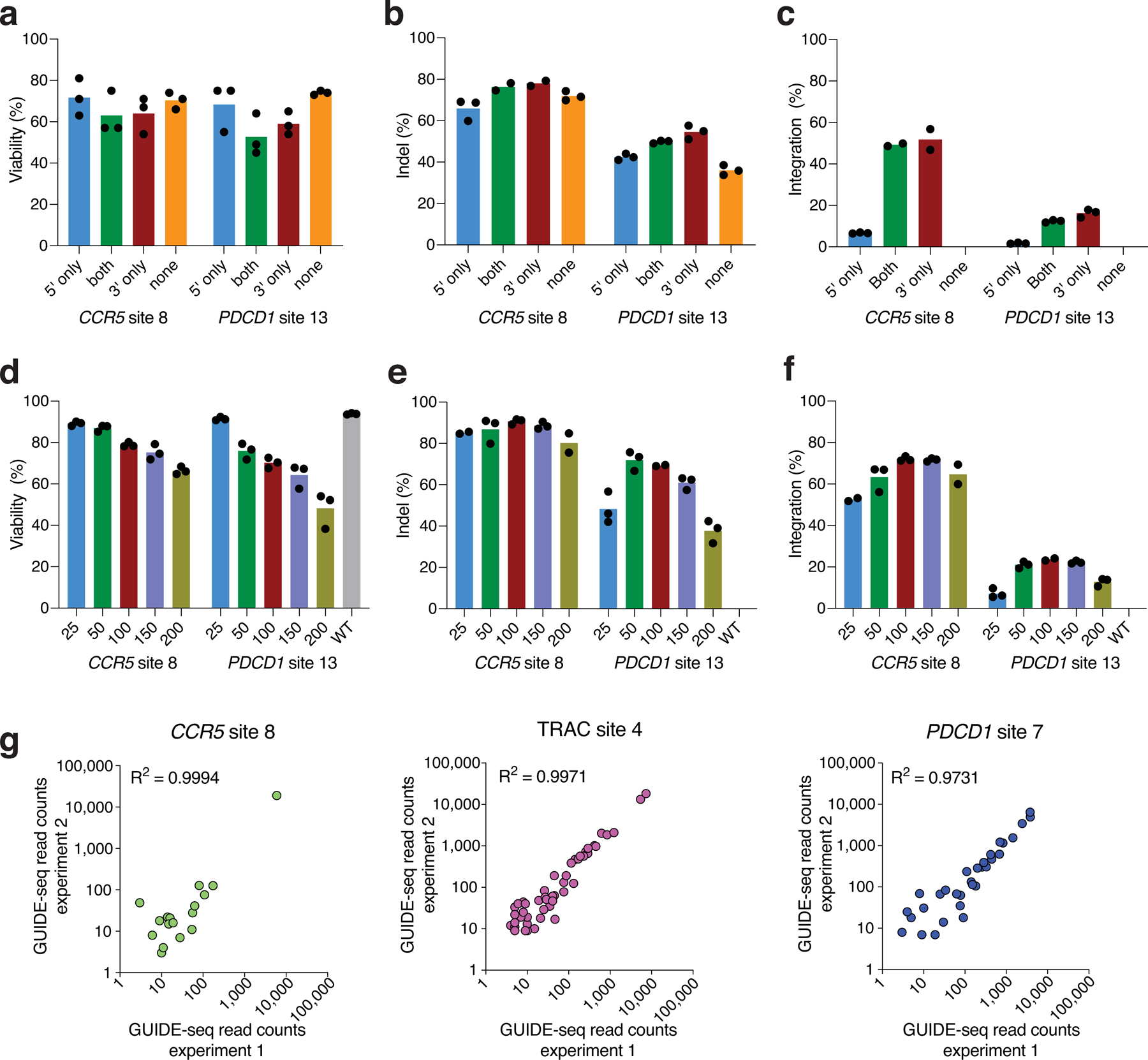
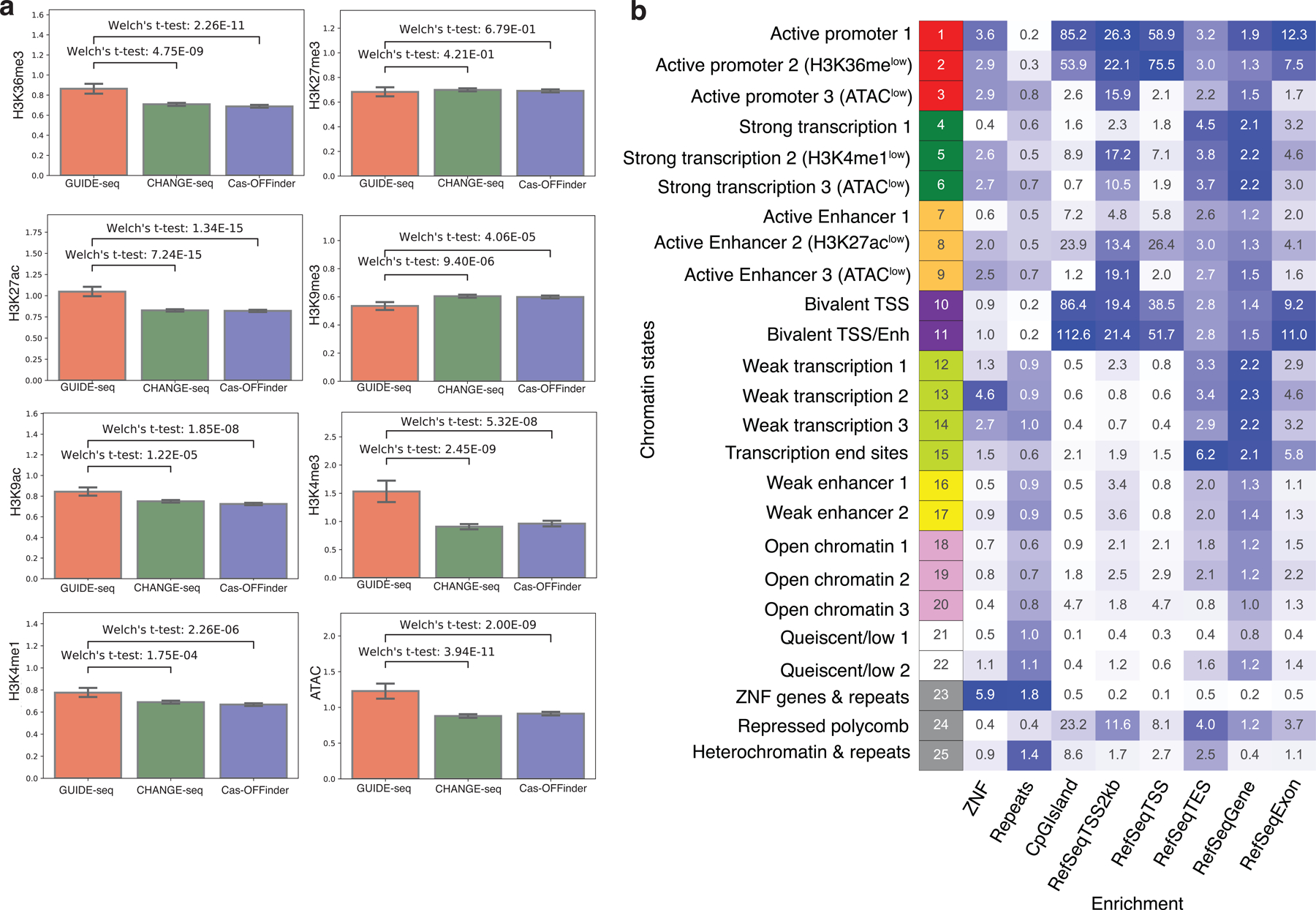
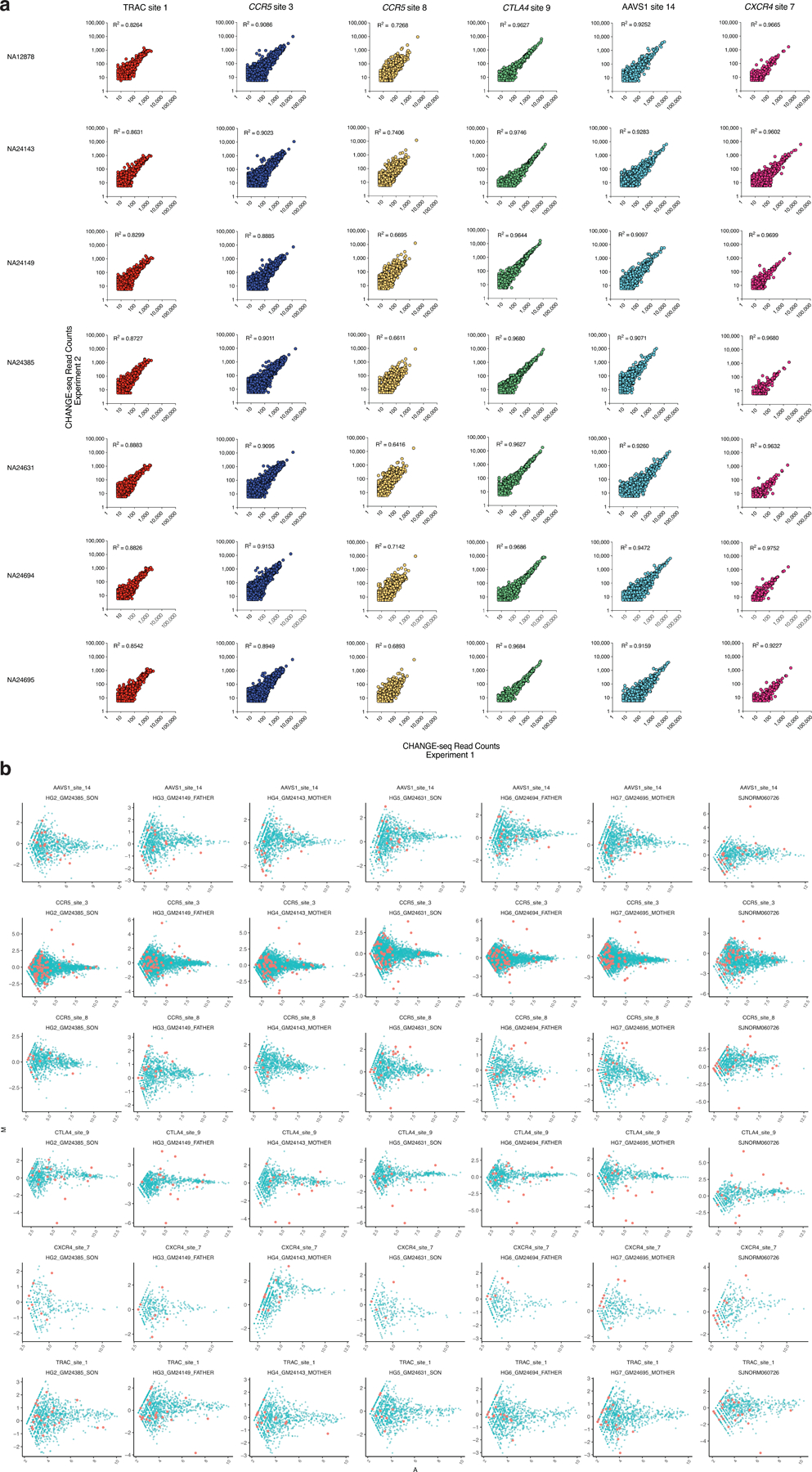
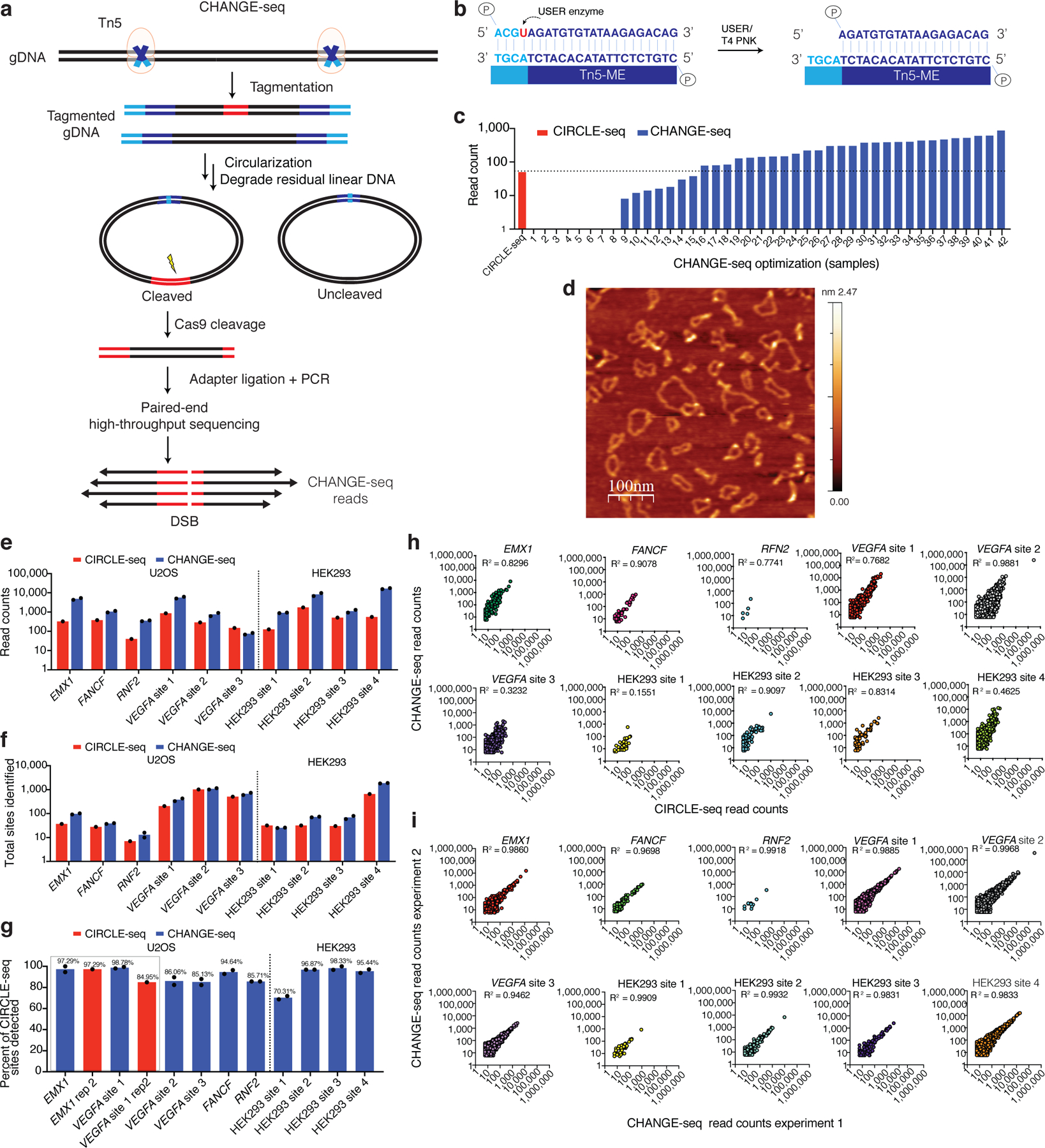
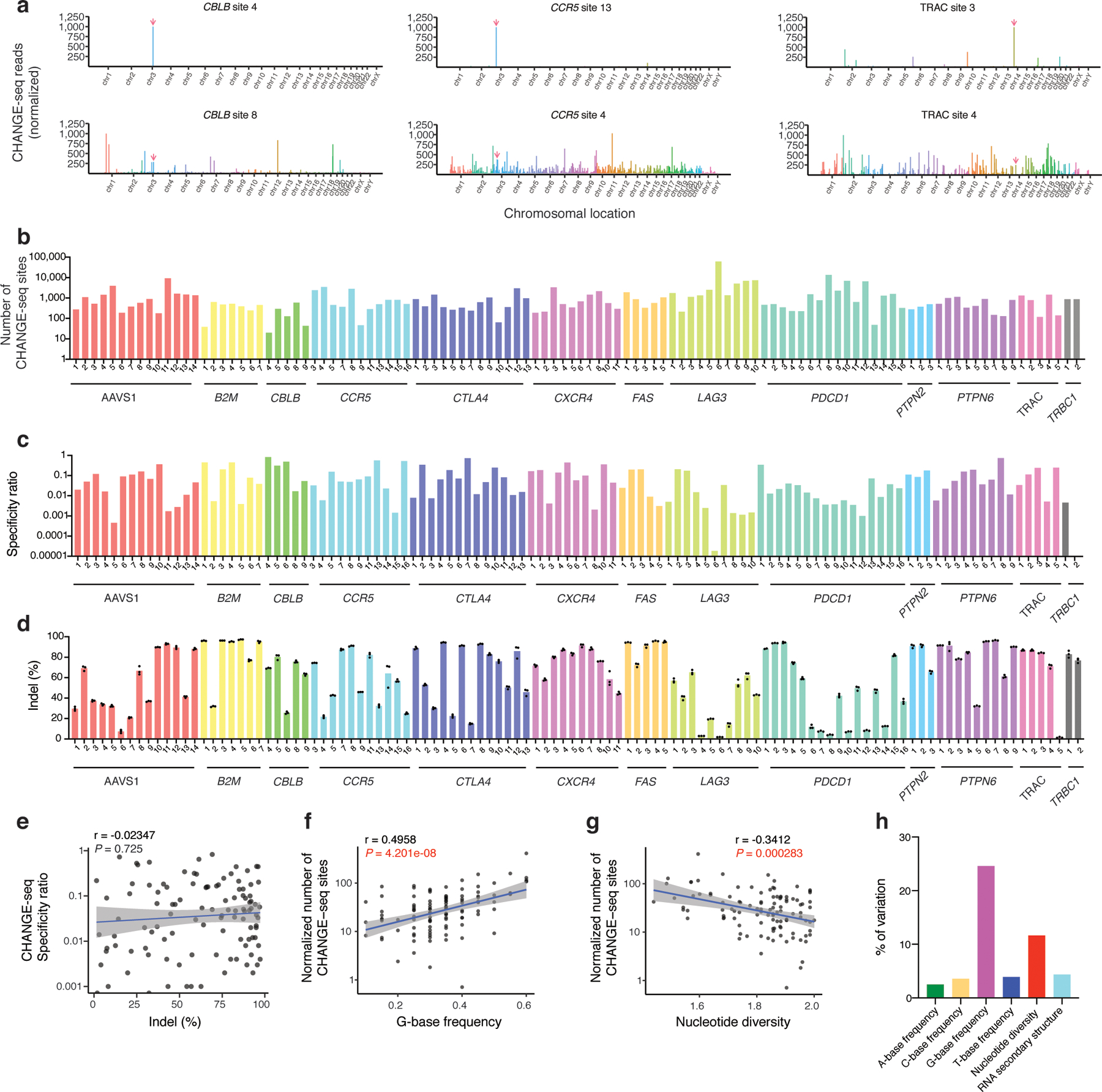
Current methods can illuminate the genome-wide activity of CRISPR-Cas9 nucleases, but are not easily scalable to the throughput needed to fully understand the principles that govern Cas9 specificity. Here we describe 'circularization for high-throughput analysis of nuclease genome-wide effects by sequencing' (CHANGE-seq), a scalable, automatable tagmentation-based method for measuring the genome-wide activity of Cas9 in vitro. We applied CHANGE-seq to 110 single guide RNA targets across 13 therapeutically relevant loci in human primary T cells and identified 201,934 off-target sites, enabling the training of a machine learning model to predict off-target activity. Comparing matched genome-wide off-target, chromatin modification and accessibility, and transcriptional data, we found that cellular off-target activity was two to four times more likely to occur near active promoters, enhancers and transcribed regions. Finally, CHANGE-seq analysis of six targets across eight individual genomes revealed that human single-nucleotide variation had significant effects on activity at ~15.2% of off-target sites analyzed. CHANGE-seq is a simplified, sensitive and scalable approach to understanding the specificity of genome editors.
Conflict of interest statement
Competing Interests Statement
C.R.L. and S.Q.T. have filed a patent application on CHANGE-seq. S.Q.T. is a co-inventor on patents covering CIRCLE-seq and GUIDE-seq. S.Q.T. is a member of the scientific advisory board of Kromatid.
Figures
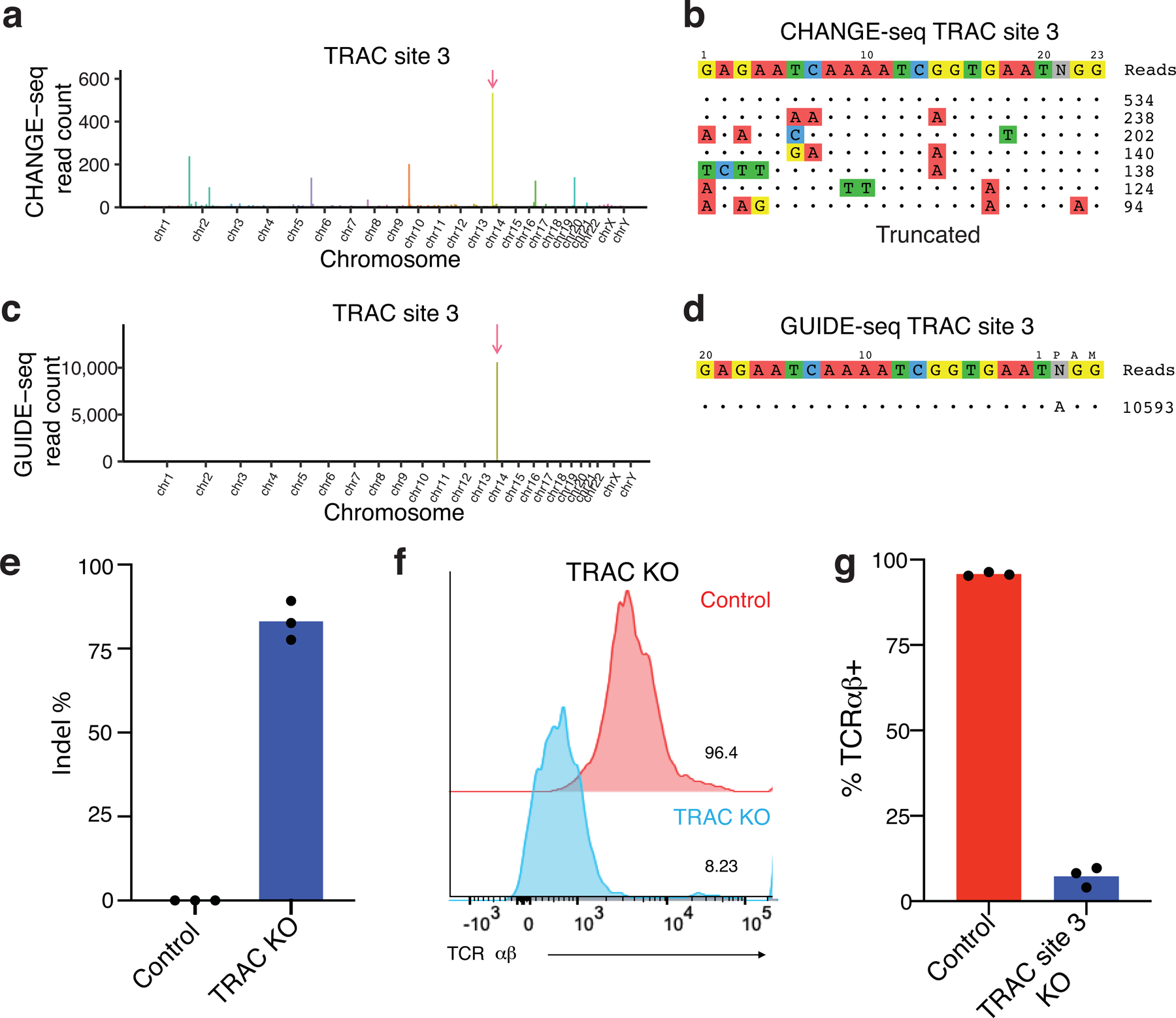
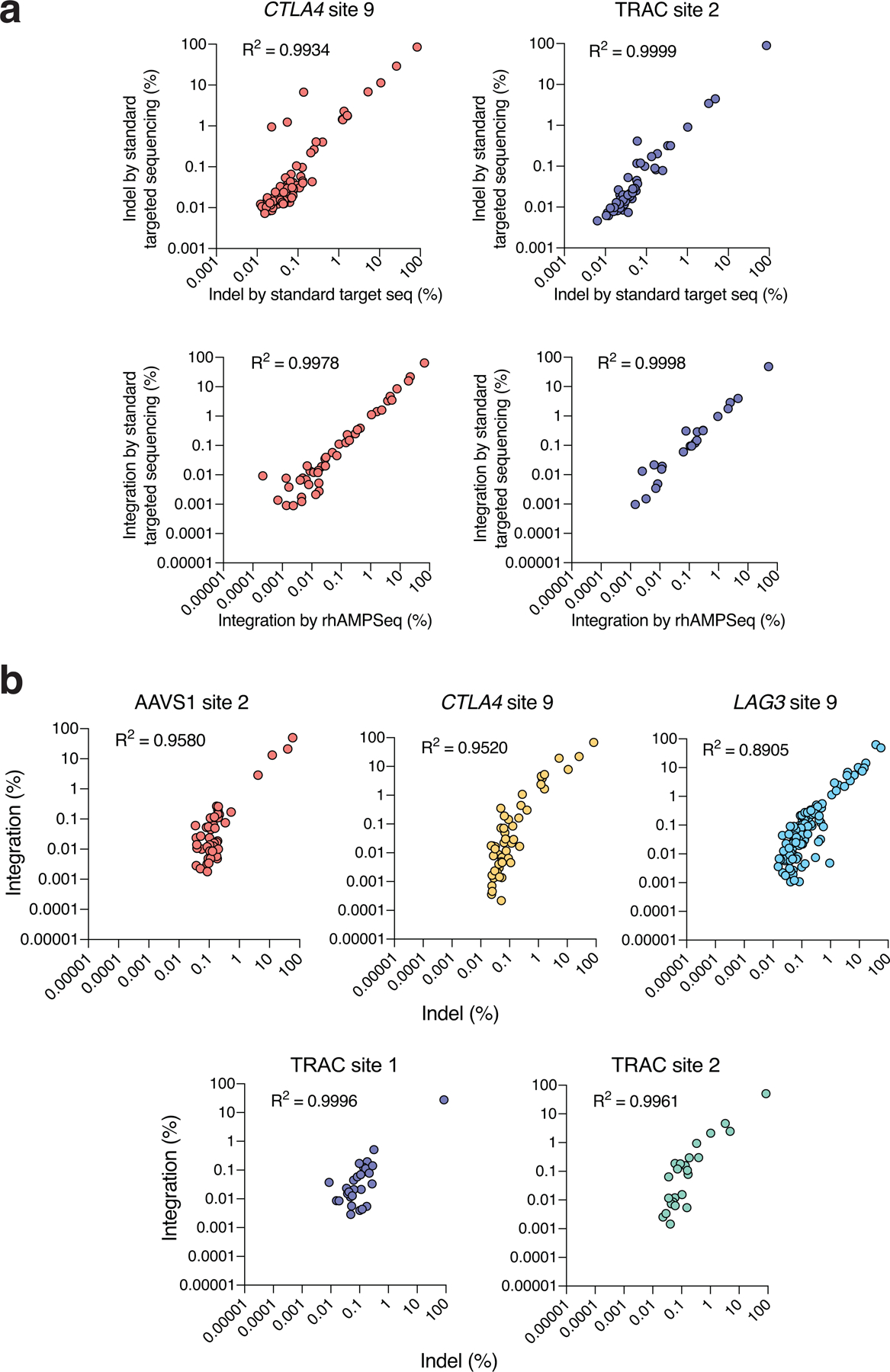
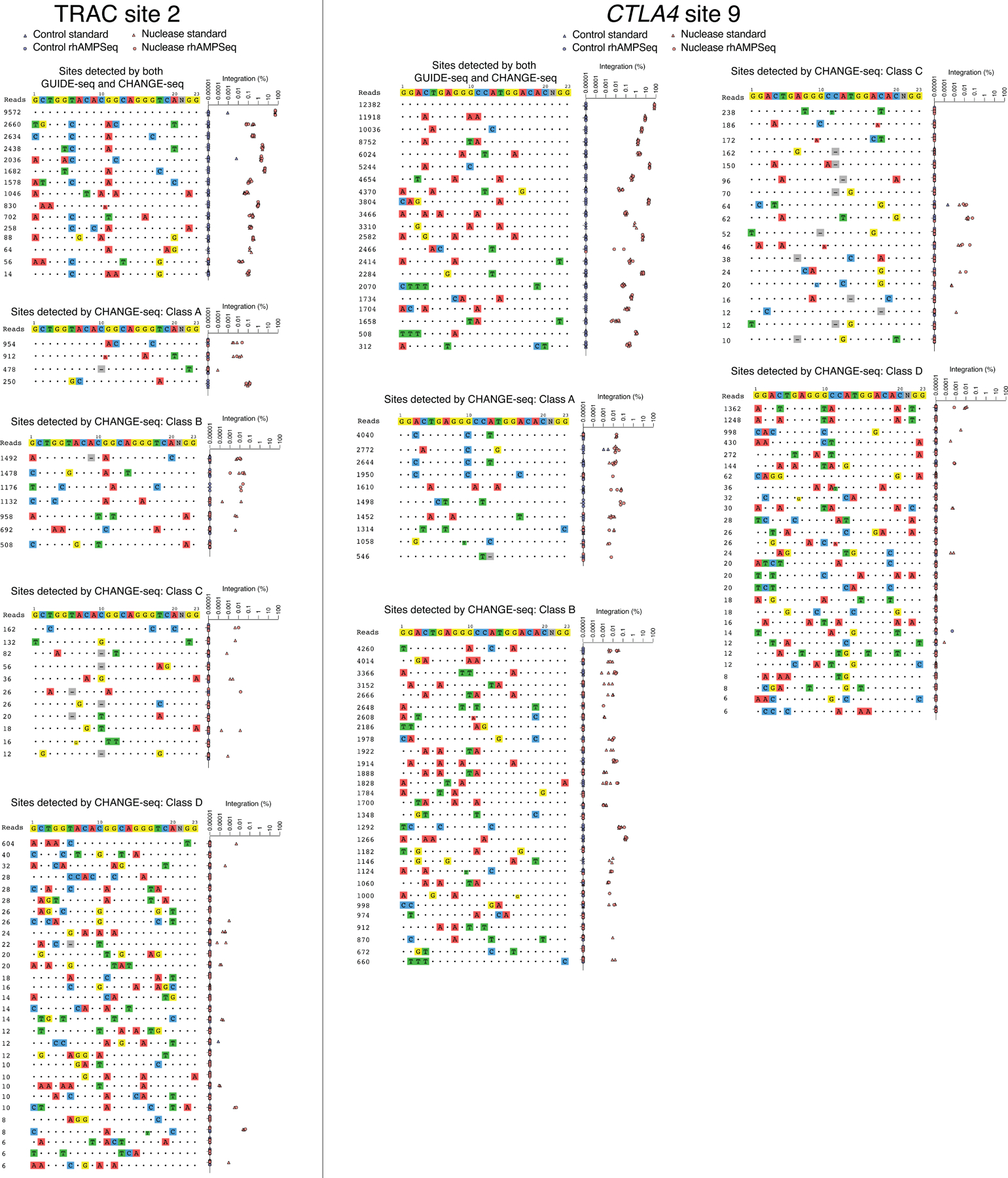
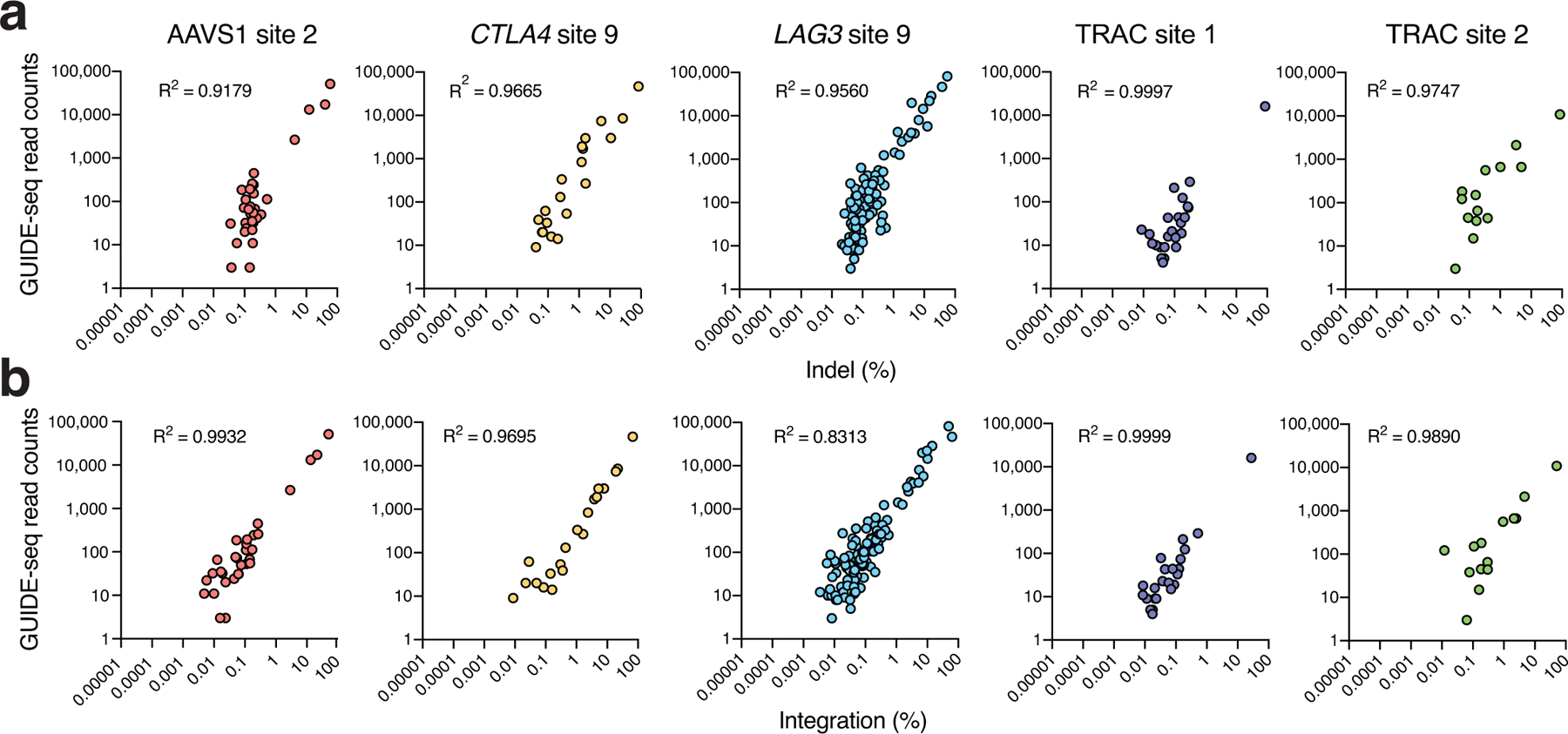
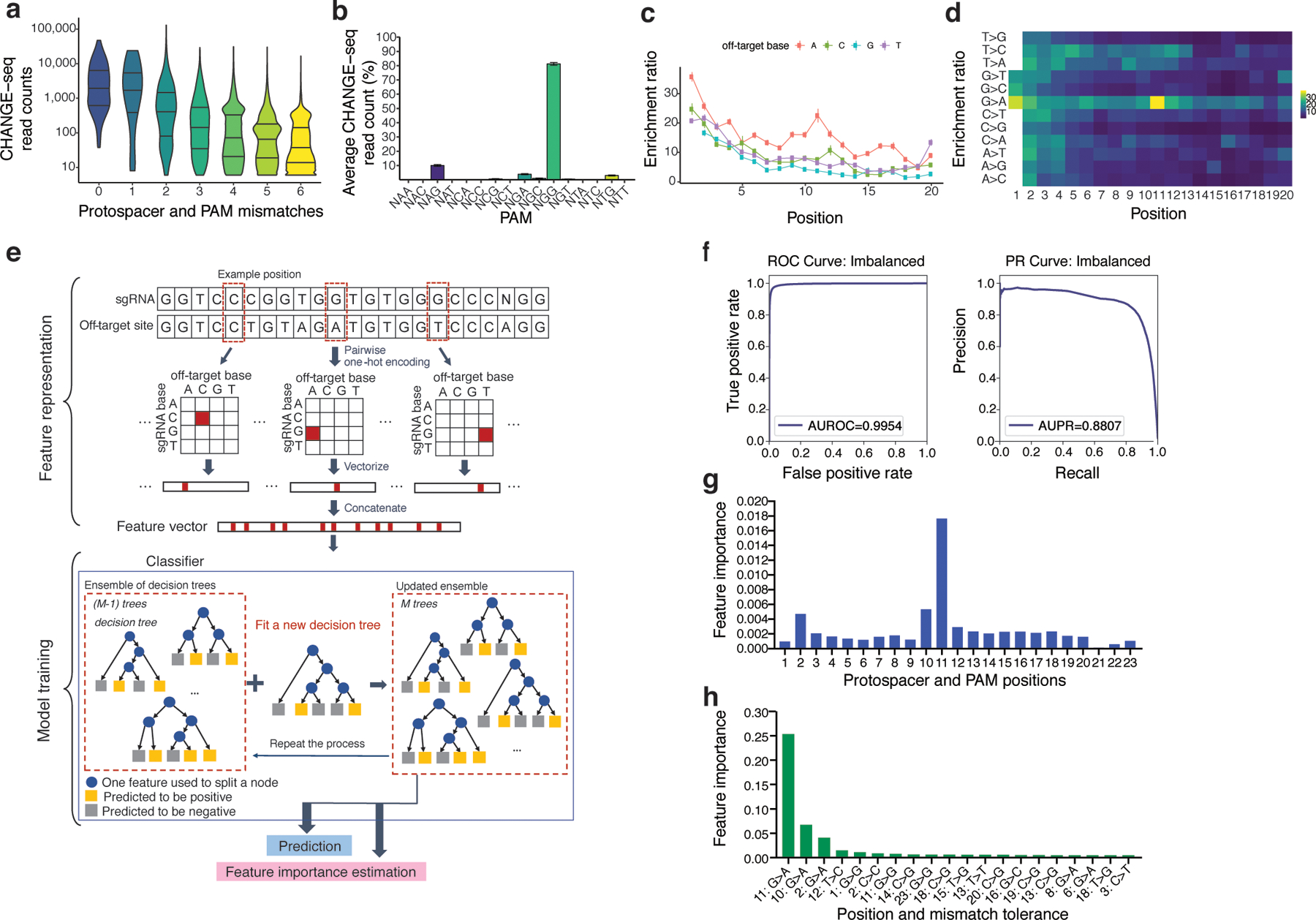
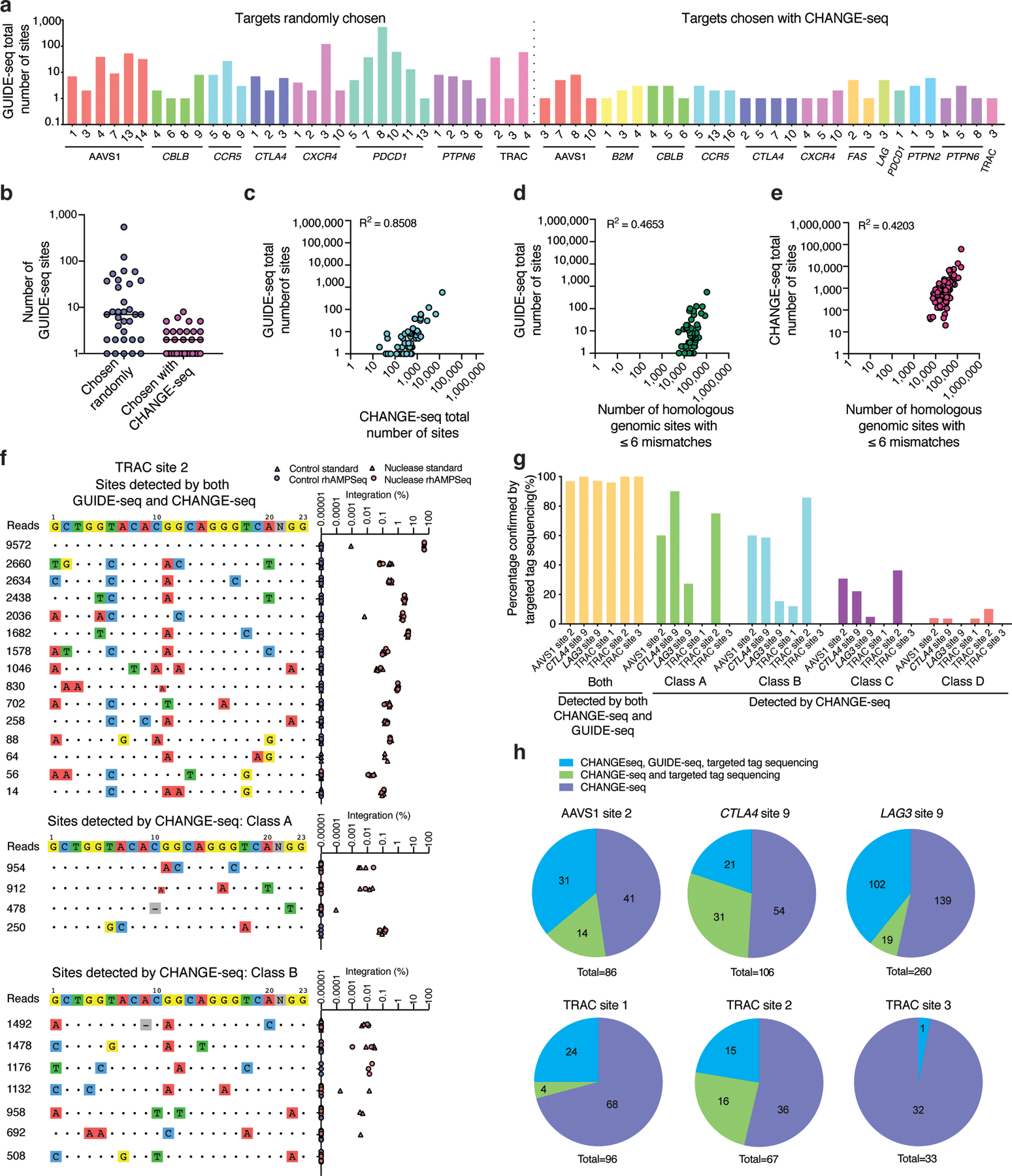
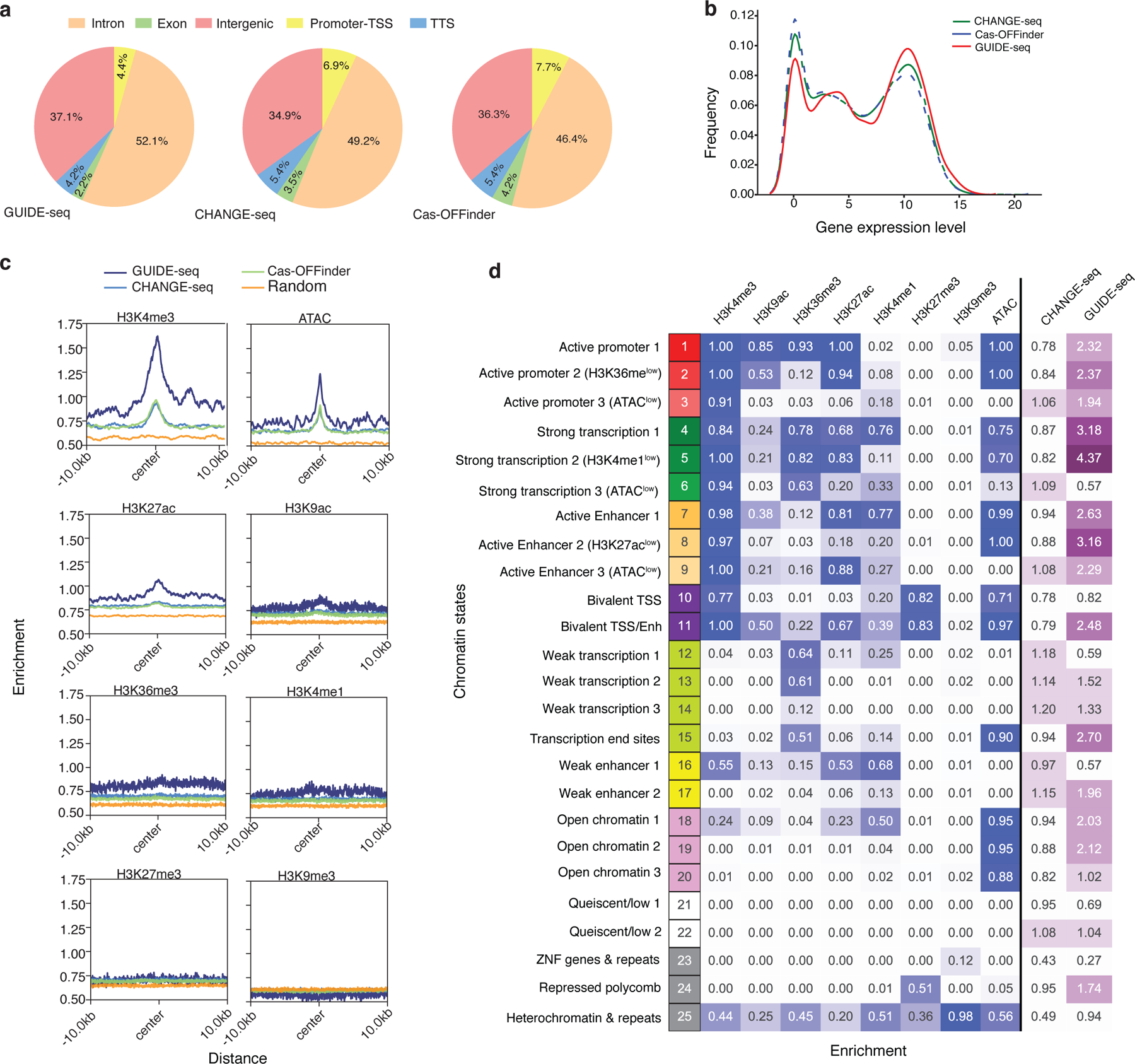
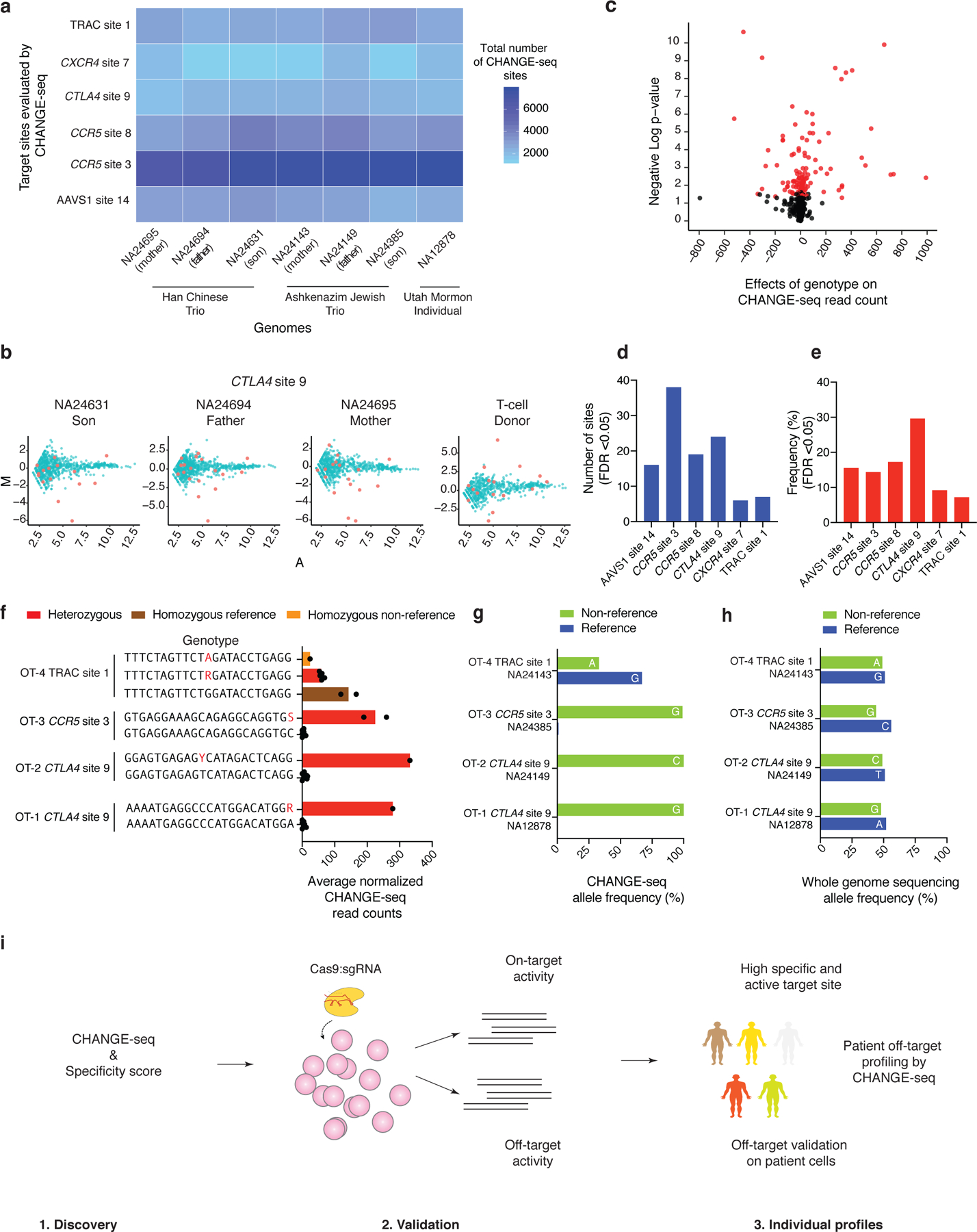
References
-
- Hacein-Bey-Abina S et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 302, 415–419 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
