

Autoimmune Pathology in Myasthenia Gravis Disease Subtypes Is Governed by Divergent Mechanisms of Immunopathology
- PMID: 32547535
- PMCID: PMC7274207
- DOI: 10.3389/fimmu.2020.00776
Autoimmune Pathology in Myasthenia Gravis Disease Subtypes Is Governed by Divergent Mechanisms of Immunopathology
Abstract
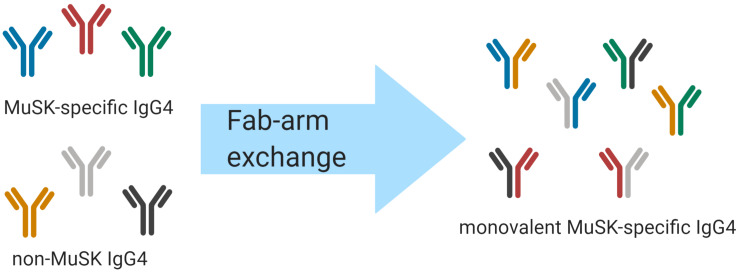
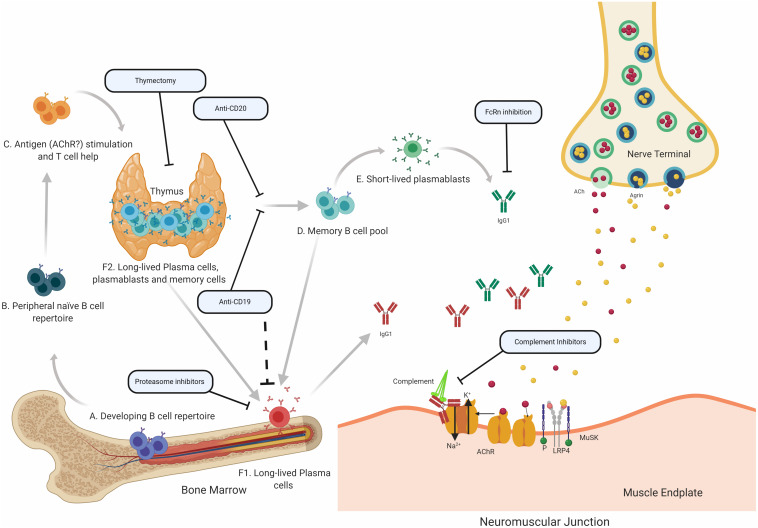
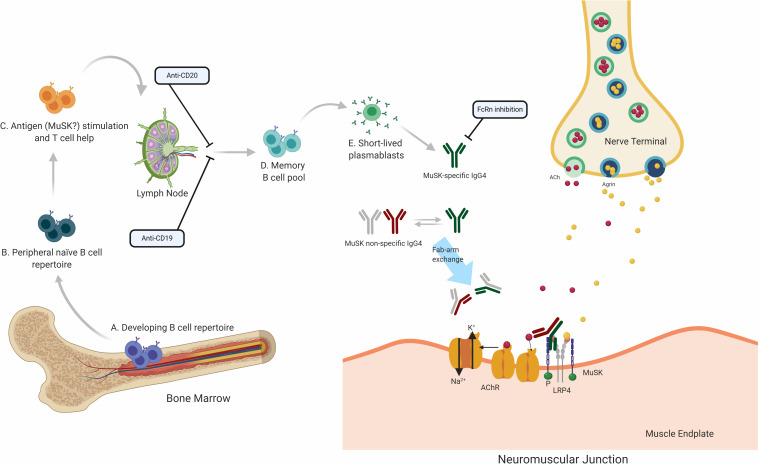
Myasthenia gravis (MG) is a prototypical autoantibody mediated disease. The autoantibodies in MG target structures within the neuromuscular junction (NMJ), thus affecting neuromuscular transmission. The major disease subtypes of autoimmune MG are defined by their antigenic target. The most common target of pathogenic autoantibodies in MG is the nicotinic acetylcholine receptor (AChR), followed by muscle-specific kinase (MuSK) and lipoprotein receptor-related protein 4 (LRP4). MG patients present with similar symptoms independent of the underlying subtype of disease, while the immunopathology is remarkably distinct. Here we highlight these distinct immune mechanisms that describe both the B cell- and autoantibody-mediated pathogenesis by comparing AChR and MuSK MG subtypes. In our discussion of the AChR subtype, we focus on the role of long-lived plasma cells in the production of pathogenic autoantibodies, the IgG1 subclass mediated pathology, and contributions of complement. The similarities underlying the immunopathology of AChR MG and neuromyelitis optica (NMO) are highlighted. In contrast, MuSK MG is caused by autoantibody production by short-lived plasmablasts. MuSK MG autoantibodies are mainly of the IgG4 subclass which can undergo Fab-arm exchange (FAE), a process unique to this subclass. In FAE IgG4, molecules can dissociate into two halves and recombine with other half IgG4 molecules resulting in bispecific antibodies. Similarities between MuSK MG and other IgG4-mediated autoimmune diseases, including pemphigus vulgaris (PV) and chronic inflammatory demyelinating polyneuropathy (CIDP), are highlighted. Finally, the immunological distinctions are emphasized through presentation of biological therapeutics that provide clinical benefit depending on the MG disease subtype.
Keywords: MuSK; AChR; B cells; B lymphocytes; autoantibodies; autoimmunity; immunopathology; myasthenia gravis.
Copyright © 2020 Fichtner, Jiang, Bourke, Nowak and O’Connor.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous