

Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy
- PMID: 32548275
- PMCID: PMC7251510
- DOI: 10.1212/NXG.0000000000000428
Mutations in the m-AAA proteases AFG3L2 and SPG7 are causing isolated dominant optic atrophy
Abstract
Objective: To improve the genetic diagnosis of dominant optic atrophy (DOA), the most frequently inherited optic nerve disease, and infer genotype-phenotype correlations.
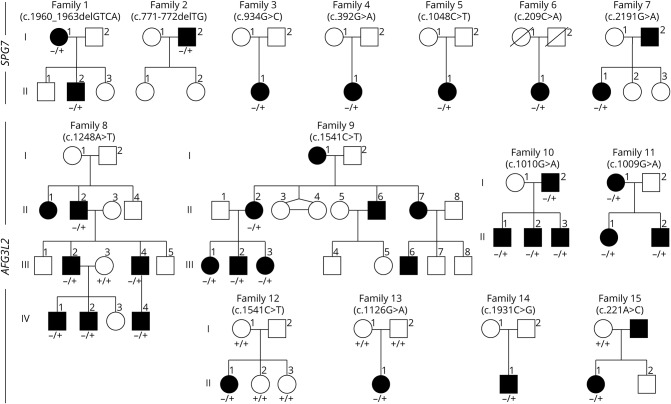
Methods: Exonic sequences of 22 genes were screened by new-generation sequencing in patients with DOA who were investigated for ophthalmology, neurology, and brain MRI.
Results: We identified 7 and 8 new heterozygous pathogenic variants in SPG7 and AFG3L2. Both genes encode for mitochondrial matricial AAA (m-AAA) proteases, initially involved in recessive hereditary spastic paraplegia type 7 (HSP7) and dominant spinocerebellar ataxia 28 (SCA28), respectively. Notably, variants in AFG3L2 that result in DOA are located in different domains to those reported in SCA28, which likely explains the lack of clinical overlap between these 2 phenotypic manifestations. In comparison, the SPG7 variants identified in DOA are interspersed among those responsible for HSP7 in which optic neuropathy has previously been reported.
Conclusions: Our results position SPG7 and AFG3L2 as candidate genes to be screened in DOA and indicate that regulation of mitochondrial protein homeostasis and maturation by m-AAA proteases are crucial for the maintenance of optic nerve physiology.
Copyright © 2020 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures


References
-
- Alexander C, Votruba M, Pesch UE, et al. . OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 2000;26:211–215. - PubMed
-
- Delettre C, Lenaers G, Griffoin JM, et al. . Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000;26:207–210. - PubMed
-
- Ferré M, Caignard A, Milea D, et al. . Improved locus-specific database for OPA1 mutations allows inclusion of advanced clinical data. Hum Mutat 2015;36:20–25. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases