

Inflammasome activation and regulation: toward a better understanding of complex mechanisms
- PMID: 32550001
- PMCID: PMC7280307
- DOI: 10.1038/s41421-020-0167-x
Inflammasome activation and regulation: toward a better understanding of complex mechanisms
Abstract
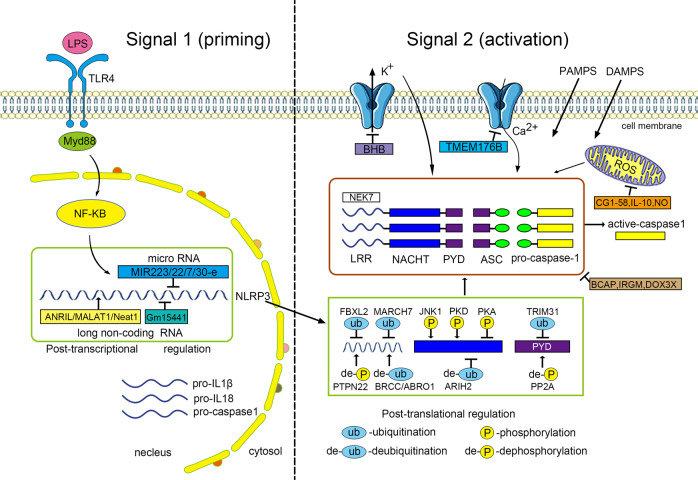
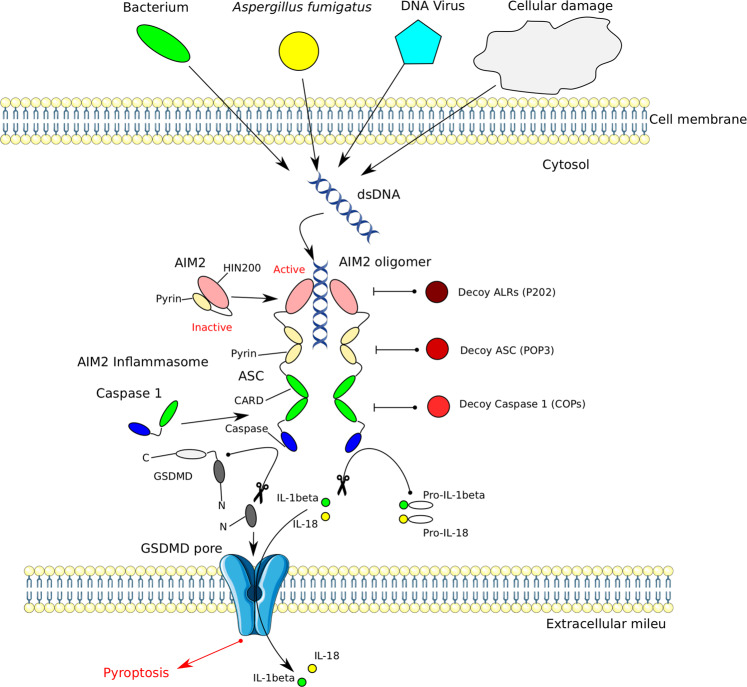
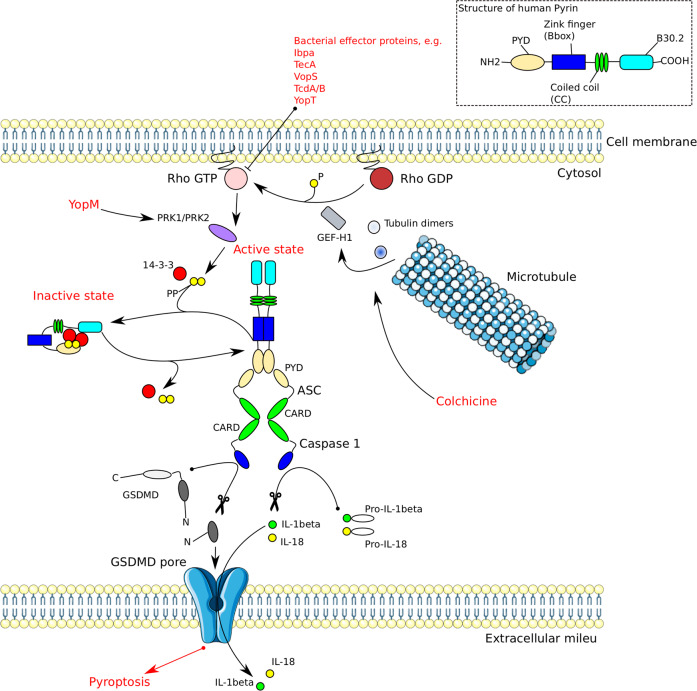
Inflammasomes are cytoplasmic multiprotein complexes comprising a sensor protein, inflammatory caspases, and in some but not all cases an adapter protein connecting the two. They can be activated by a repertoire of endogenous and exogenous stimuli, leading to enzymatic activation of canonical caspase-1, noncanonical caspase-11 (or the equivalent caspase-4 and caspase-5 in humans) or caspase-8, resulting in secretion of IL-1β and IL-18, as well as apoptotic and pyroptotic cell death. Appropriate inflammasome activation is vital for the host to cope with foreign pathogens or tissue damage, while aberrant inflammasome activation can cause uncontrolled tissue responses that may contribute to various diseases, including autoinflammatory disorders, cardiometabolic diseases, cancer and neurodegenerative diseases. Therefore, it is imperative to maintain a fine balance between inflammasome activation and inhibition, which requires a fine-tuned regulation of inflammasome assembly and effector function. Recently, a growing body of studies have been focusing on delineating the structural and molecular mechanisms underlying the regulation of inflammasome signaling. In the present review, we summarize the most recent advances and remaining challenges in understanding the ordered inflammasome assembly and activation upon sensing of diverse stimuli, as well as the tight regulations of these processes. Furthermore, we review recent progress and challenges in translating inflammasome research into therapeutic tools, aimed at modifying inflammasome-regulated human diseases.
Keywords: Cell signalling; NOD-like receptors.
© The Author(s) 2020.
Conflict of interest statement
Conflict of interestThe authors declare that they have no conflict of interest.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
