

Therapeutic potential of a TrkB agonistic antibody for Alzheimer's disease
- PMID: 32550908
- PMCID: PMC7295064
- DOI: 10.7150/thno.44165
Therapeutic potential of a TrkB agonistic antibody for Alzheimer's disease
Abstract
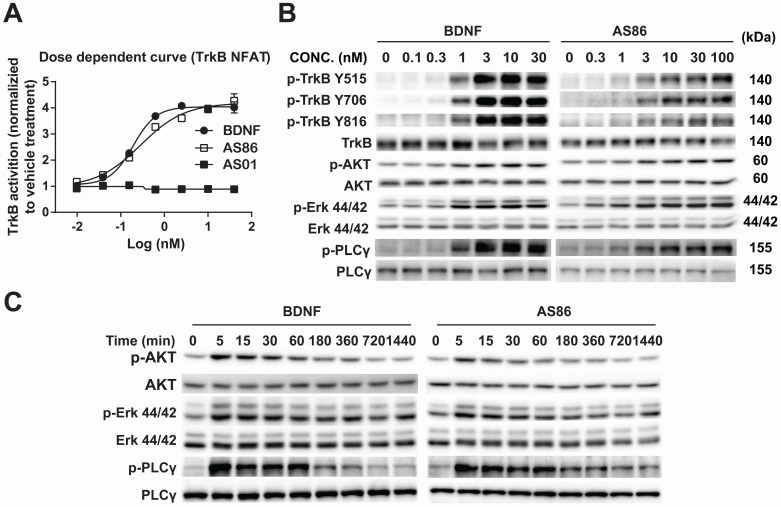
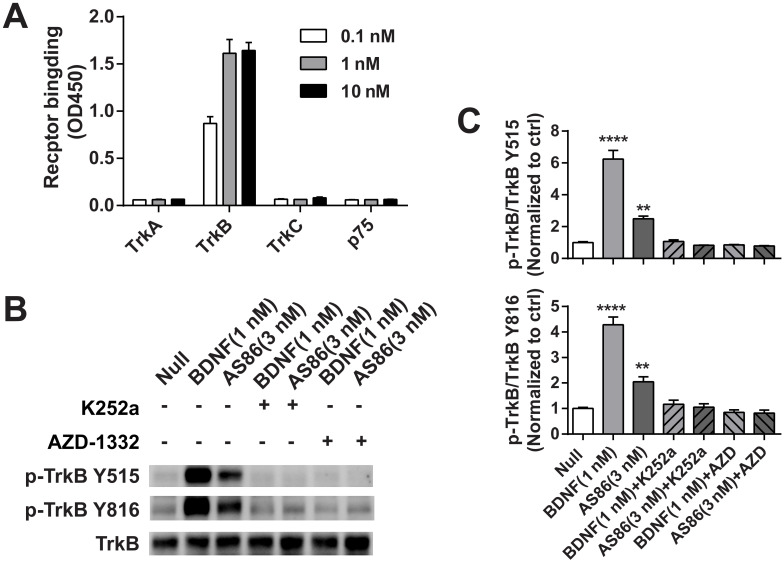
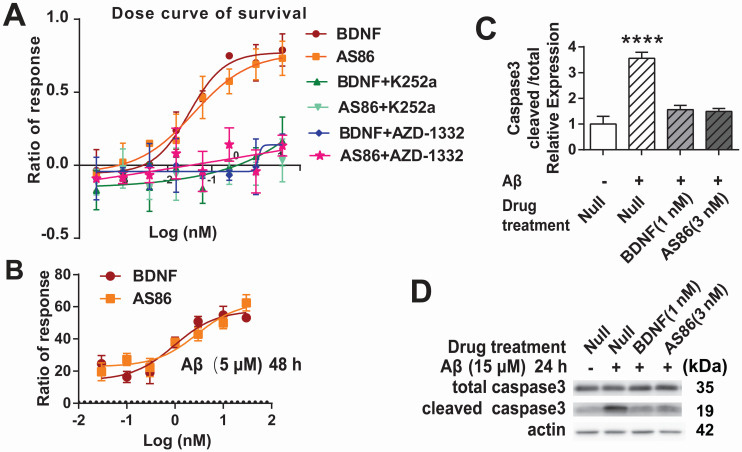
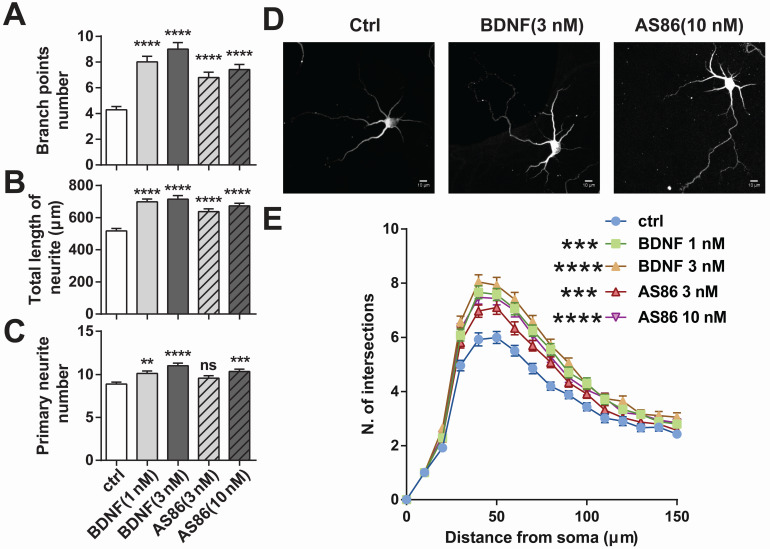
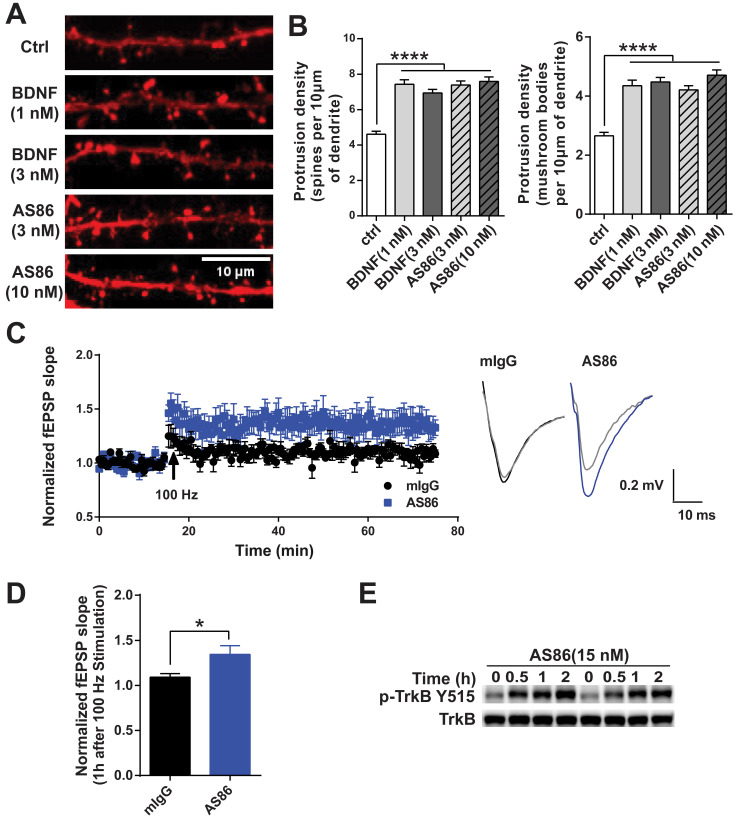
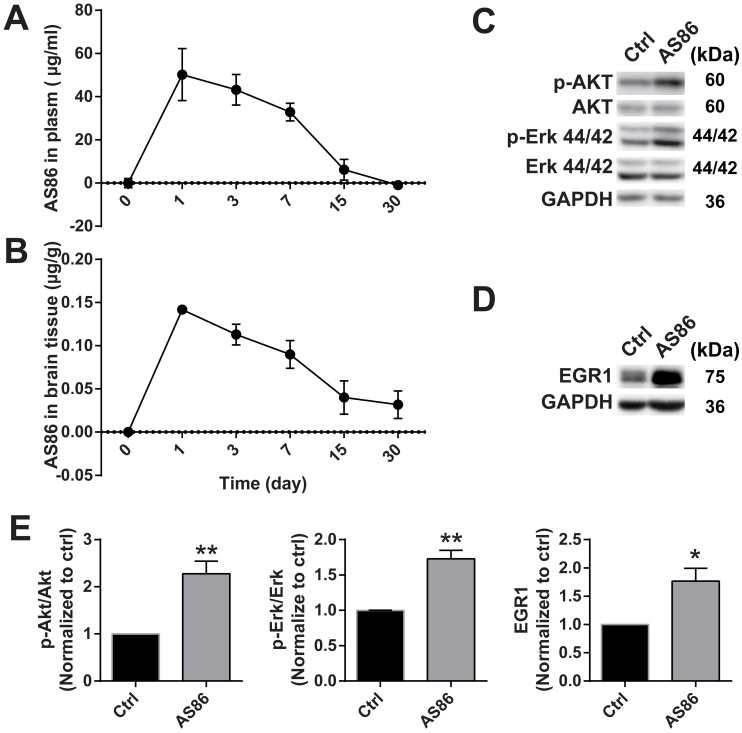
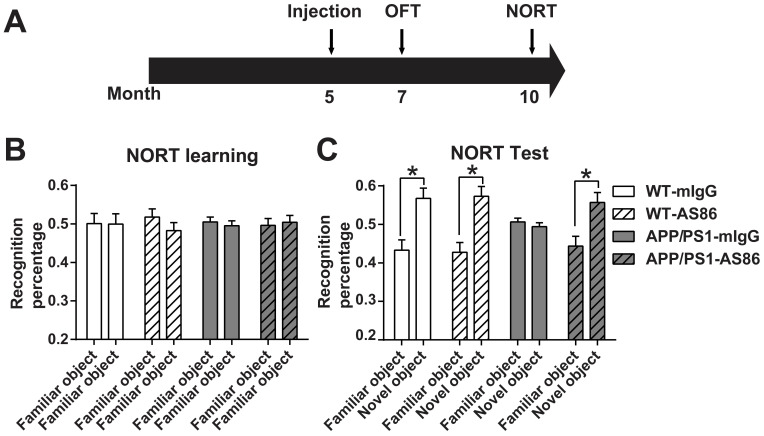
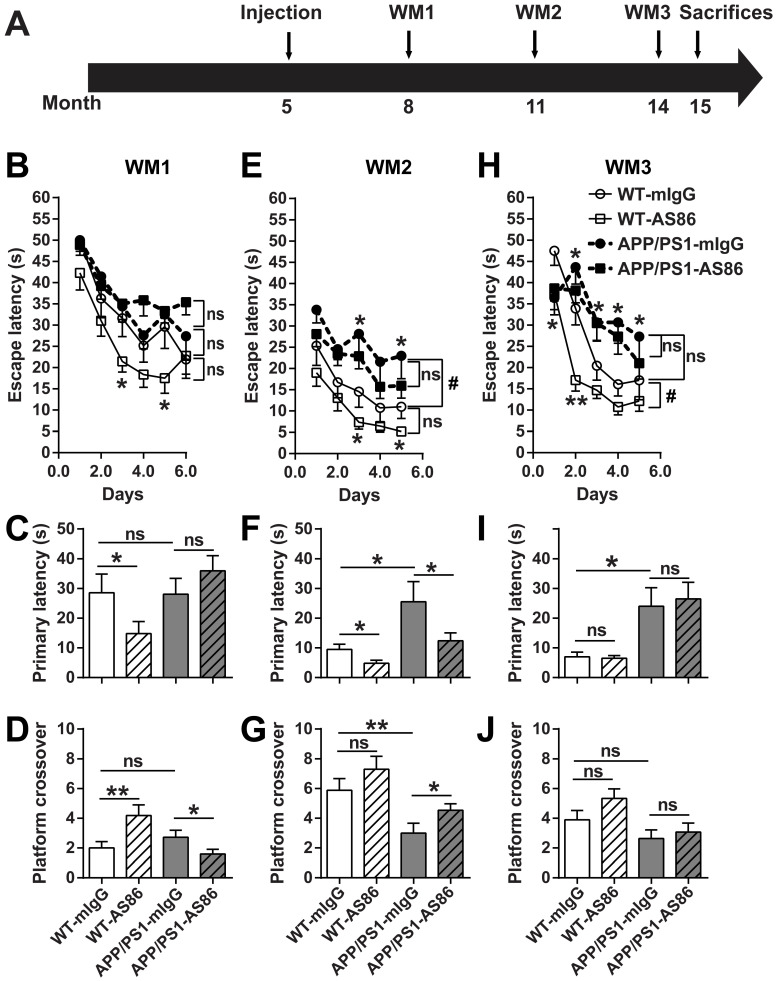
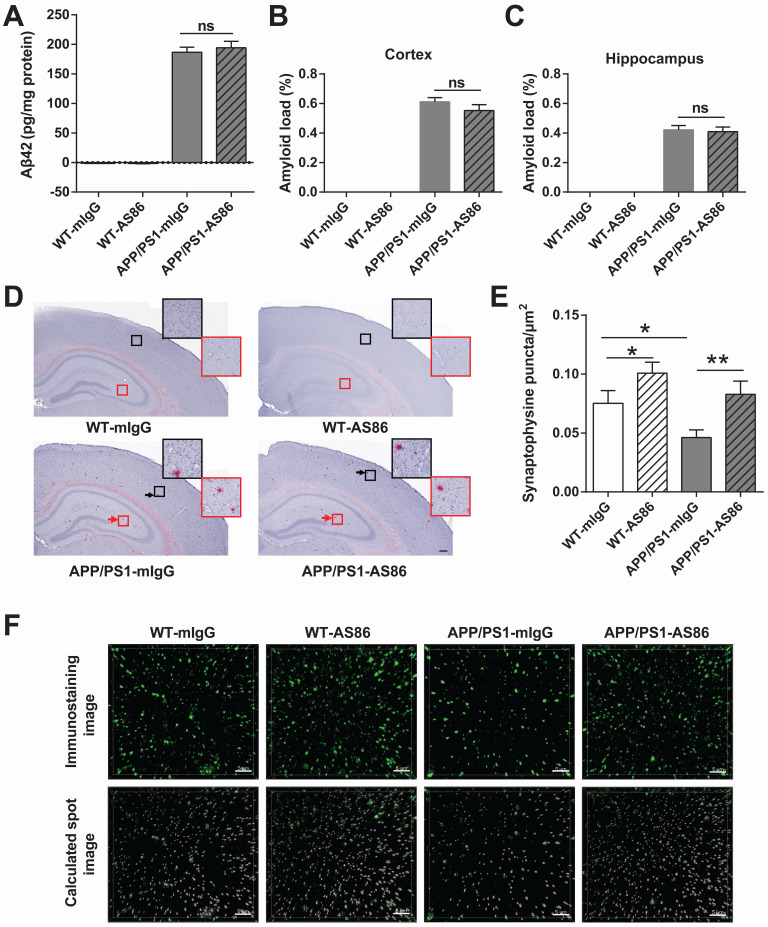
Repeated failures of "Aβ-lowering" therapies call for new targets and therapeutic approaches for Alzheimer's disease (AD). We propose to treat AD by halting neuronal death and repairing synapses using a BDNF-based therapy. To overcome the poor druggability of BDNF, we have developed an agonistic antibody AS86 to mimic the function of BDNF, and evaluate its therapeutic potential for AD. Method: Biochemical, electrophysiological and behavioral techniques were used to investigate the effects of AS86 in vitro and in vivo. Results: AS86 specifically activated the BDNF receptor TrkB and its downstream signaling, without affecting its other receptor p75NTR. It promoted neurite outgrowth, enhanced spine growth and prevented Aβ-induced cell death in cultured neurons, and facilitated Long-Term Potentiation (LTP) in hippocampal slices. A single-dose tail-vein injection of AS86 activated TrkB signaling in the brain, with a half-life of 6 days in the blood and brain. Bi-weekly peripheral administration of AS86 rescued the deficits in object-recognition memory in the APP/PS1 mouse model. AS86 also reversed spatial memory deficits in the 11-month, but not 14-month old AD mouse model. Conclusion: These results demonstrate the potential of AS86 in AD therapy, suggesting that neuronal and/or synaptic repair as an alternative therapeutic strategy for AD.
Keywords: Antibody drug; Cognition; Neurodegeneration; Synaptic plasticity; Therapy.
© The author(s).
Conflict of interest statement
Competing Interests: BL, WG and HY are co-inventors of the filed patents related to the TrkB agonistic antibodies. The remaining authors declare no conflict of interest.
Figures
Similar articles
-
A TrkB and TrkC partial agonist restores deficits in synaptic function and promotes activity-dependent synaptic and microglial transcriptomic changes in a late-stage Alzheimer's mouse model.Alzheimers Dement. 2024 Jul;20(7):4434-4460. doi: 10.1002/alz.13857. Epub 2024 May 23. Alzheimers Dement. 2024. PMID: 38779814 Free PMC article.
-
LMDS-1, a potential TrkB receptor agonist provides a safe and neurotrophic effect for early-phase Alzheimer's disease.Psychopharmacology (Berl). 2020 Oct;237(10):3173-3190. doi: 10.1007/s00213-020-05602-z. Epub 2020 Aug 4. Psychopharmacology (Berl). 2020. PMID: 32748031
-
The flavonoid baicalein rescues synaptic plasticity and memory deficits in a mouse model of Alzheimer's disease.Behav Brain Res. 2016 Sep 15;311:309-321. doi: 10.1016/j.bbr.2016.05.052. Epub 2016 May 24. Behav Brain Res. 2016. PMID: 27233830
-
The Seed of Zizyphus jujuba var. spinosa Attenuates Alzheimer's Disease-Associated Hippocampal Synaptic Deficits through BDNF/TrkB Signaling.Biol Pharm Bull. 2017;40(12):2096-2104. doi: 10.1248/bpb.b17-00378. Biol Pharm Bull. 2017. PMID: 29199234
-
Unlocking Alzheimer's Disease: The Role of BDNF Signaling in Neuropathology and Treatment.Neuromolecular Med. 2025 May 17;27(1):36. doi: 10.1007/s12017-025-08857-x. Neuromolecular Med. 2025. PMID: 40380033 Free PMC article. Review.
Cited by
-
The causal involvement of the BDNF-TrkB pathway in dentate gyrus in early-life stress-induced cognitive deficits in male mice.Transl Psychiatry. 2023 May 24;13(1):173. doi: 10.1038/s41398-023-02476-5. Transl Psychiatry. 2023. PMID: 37225683 Free PMC article.
-
The Role of Microglia in the Development of Neurodegenerative Diseases.Biomedicines. 2021 Oct 12;9(10):1449. doi: 10.3390/biomedicines9101449. Biomedicines. 2021. PMID: 34680566 Free PMC article. Review.
-
Pharmacological Strategies to Improve Dendritic Spines in Alzheimer's Disease.J Alzheimers Dis. 2021;82(s1):S91-S107. doi: 10.3233/JAD-201106. J Alzheimers Dis. 2021. PMID: 33325386 Free PMC article. Review.
-
Using ΔK280 TauRD Folding Reporter Cells to Screen TRKB Agonists as Alzheimer's Disease Treatment Strategy.Biomolecules. 2023 Jan 23;13(2):219. doi: 10.3390/biom13020219. Biomolecules. 2023. PMID: 36830589 Free PMC article.
-
BDNF haploinsufficiency induces behavioral endophenotypes of schizophrenia in male mice that are rescued by enriched environment.Transl Psychiatry. 2021 Apr 22;11(1):233. doi: 10.1038/s41398-021-01365-z. Transl Psychiatry. 2021. PMID: 33888685 Free PMC article.
References
-
- Polanco JC, Li C, Bodea L-G, Martinez-Marmol R, Meunier FA, Götz J. Amyloid-β and tau complexity — towards improved biomarkers and targeted therapies. Nat Rev Neurol. 2017;14:22. - PubMed
-
- Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Pri. 2015;1:15056. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
