

SMRT sequencing of the full-length transcriptome of the white-backed planthopper Sogatella furcifera
- PMID: 32551204
- PMCID: PMC7292024
- DOI: 10.7717/peerj.9320
SMRT sequencing of the full-length transcriptome of the white-backed planthopper Sogatella furcifera
Abstract
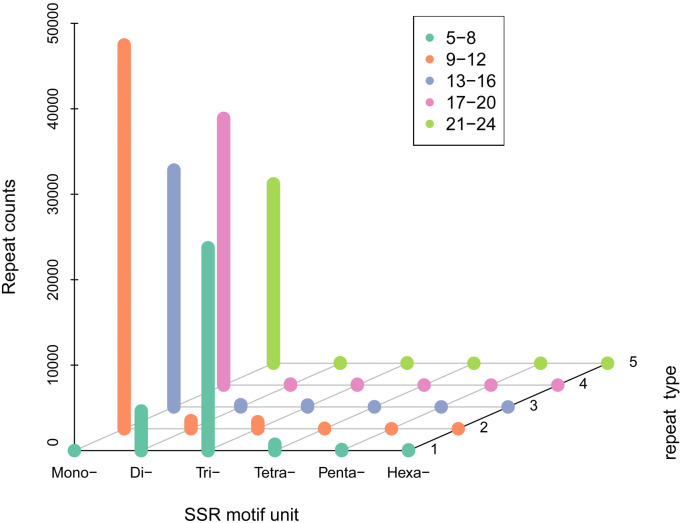
The white-backed planthopper Sogatella furcifera is an economically important rice pest distributed throughout Asia. It damages rice crops by sucking phloem sap, resulting in stunted growth and plant virus transmission. We aimed to obtain the full-length transcriptome data of S. furcifera using PacBio single-molecule real-time (SMRT) sequencing. Total RNA extracted from S. furcifera at various developmental stages (egg, larval, and adult stages) was mixed and used to generate a full-length transcriptome for SMRT sequencing. Long non-coding RNA (lncRNA) identification, full-length coding sequence prediction, full-length non-chimeric (FLNC) read detection, simple sequence repeat (SSR) analysis, transcription factor detection, and transcript functional annotation were performed. A total of 12,514,449 subreads (15.64 Gbp, clean reads) were generated, including 630,447 circular consensus sequences and 388,348 FLNC reads. Transcript cluster analysis of the FLNC reads revealed 251,109 consensus reads including 29,700 high-quality reads. Additionally, 100,360 SSRs and 121,395 coding sequences were identified using SSR analysis and ANGEL software, respectively. Furthermore, 44,324 lncRNAs were annotated using four tools and 1,288 transcription factors were identified. In total, 95,495 transcripts were functionally annotated based on searches of seven different databases. To the best of our knowledge, this is the first study of the full-length transcriptome of the white-backed planthopper obtained using SMRT sequencing. The acquired transcriptome data can facilitate further studies on the ecological and viral-host interactions of this agricultural pest.
Keywords: Full-length transcriptome; SMRT sequencing; Sogatella furcifera.
©2020 Chen et al.
Conflict of interest statement
The authors declare there are no competing interests.
Figures





References
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
