

Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato
- PMID: 32553272
- PMCID: PMC7354227
- DOI: 10.1016/j.cell.2020.05.021
Major Impacts of Widespread Structural Variation on Gene Expression and Crop Improvement in Tomato
Abstract
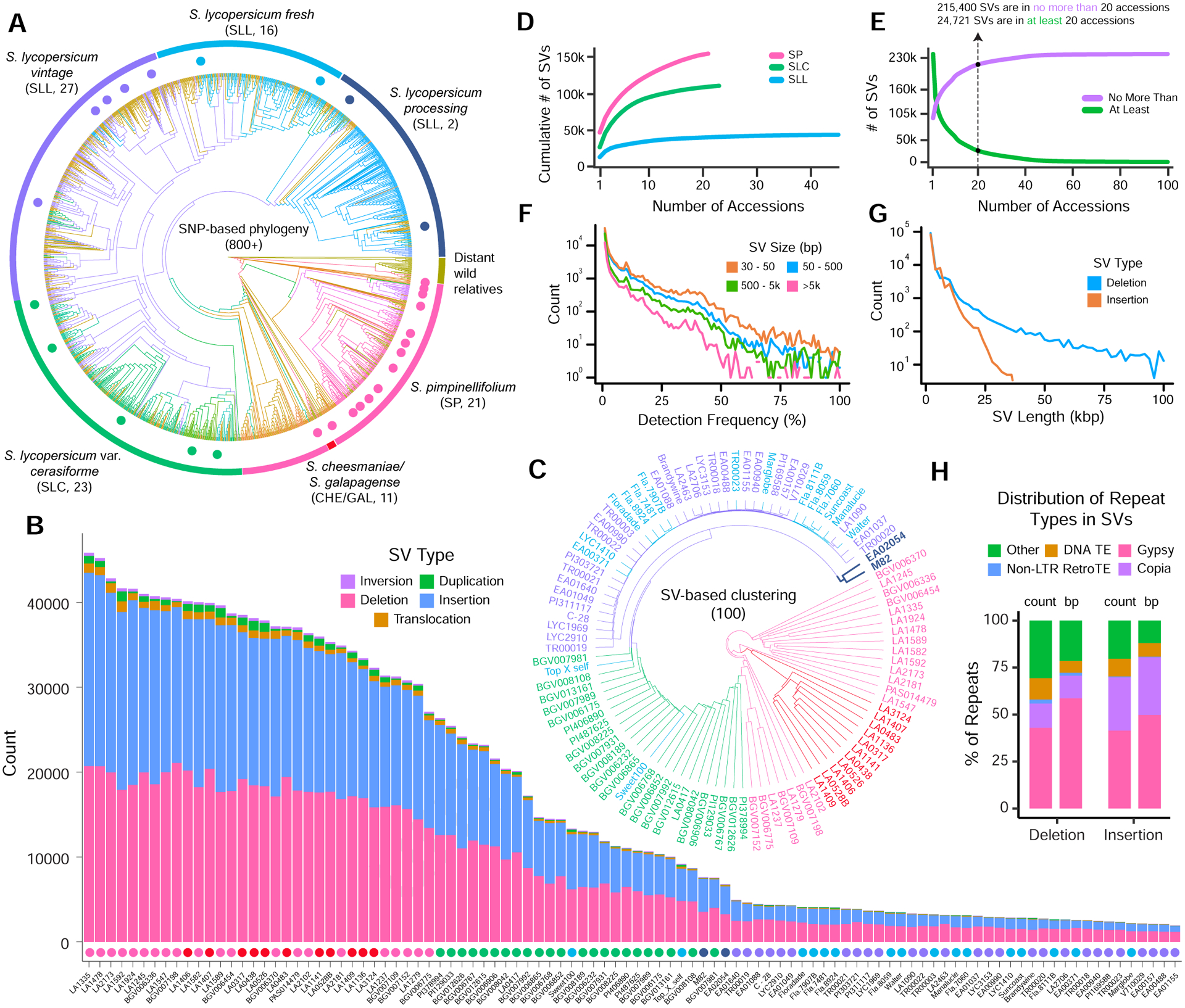
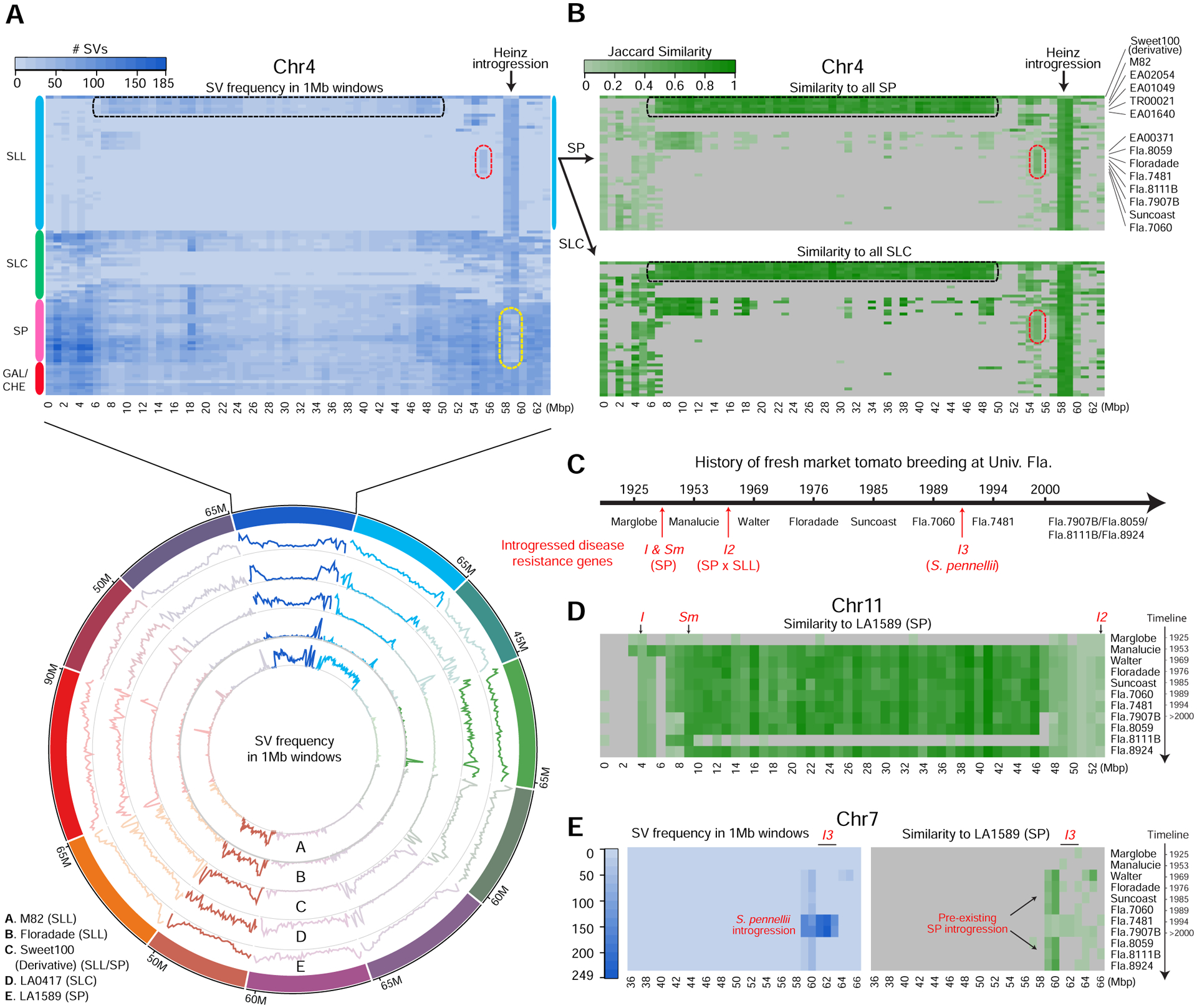
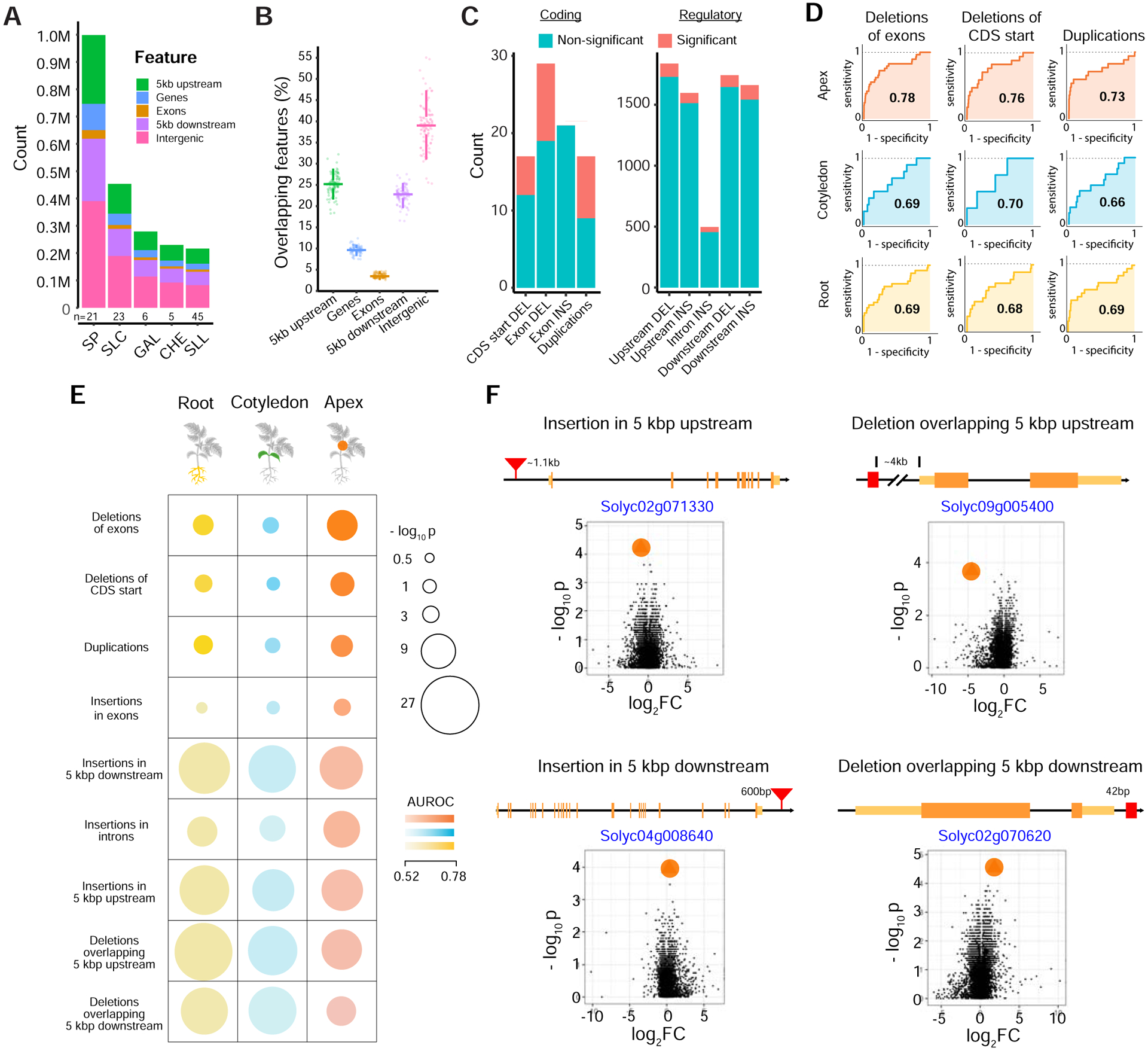
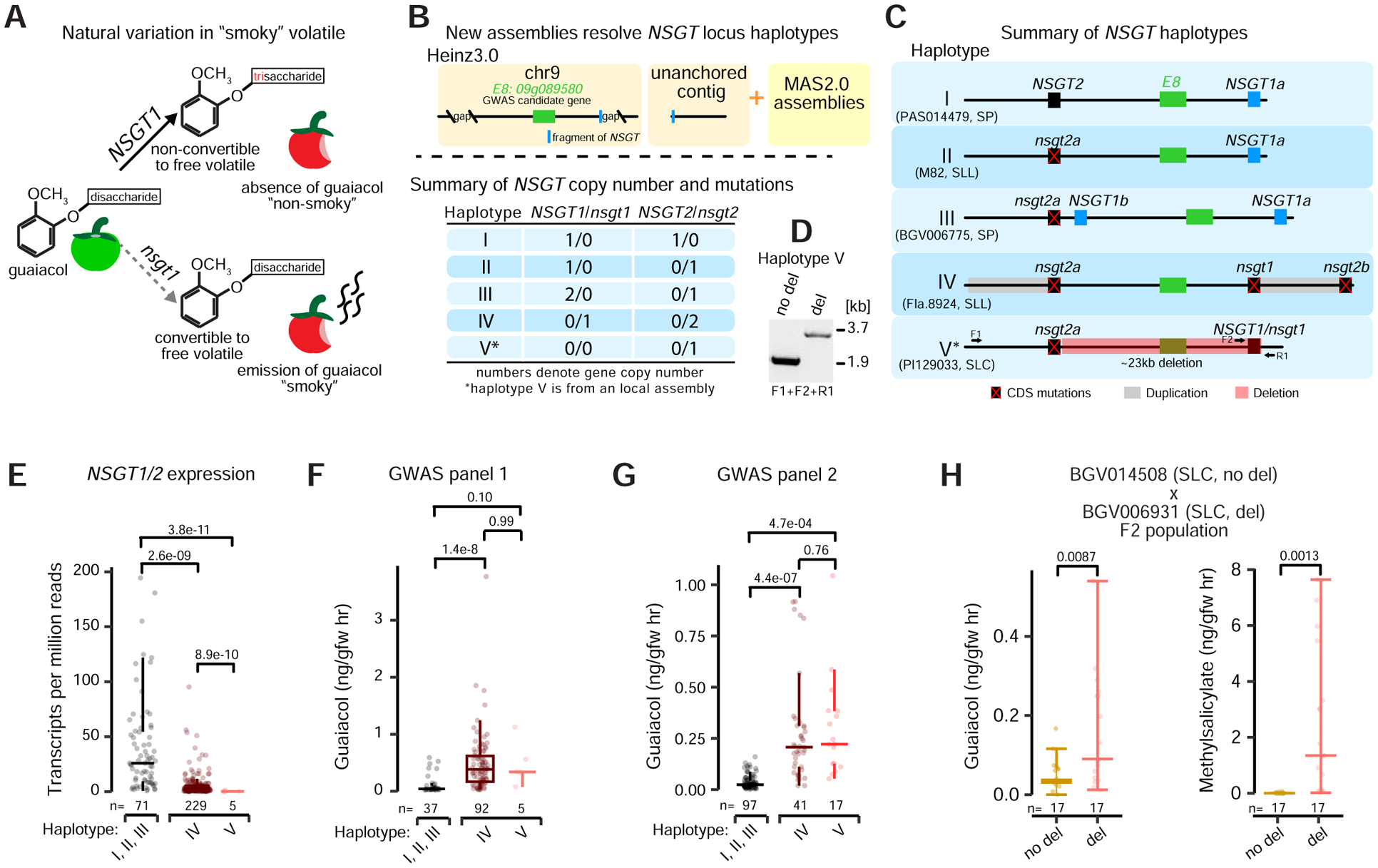
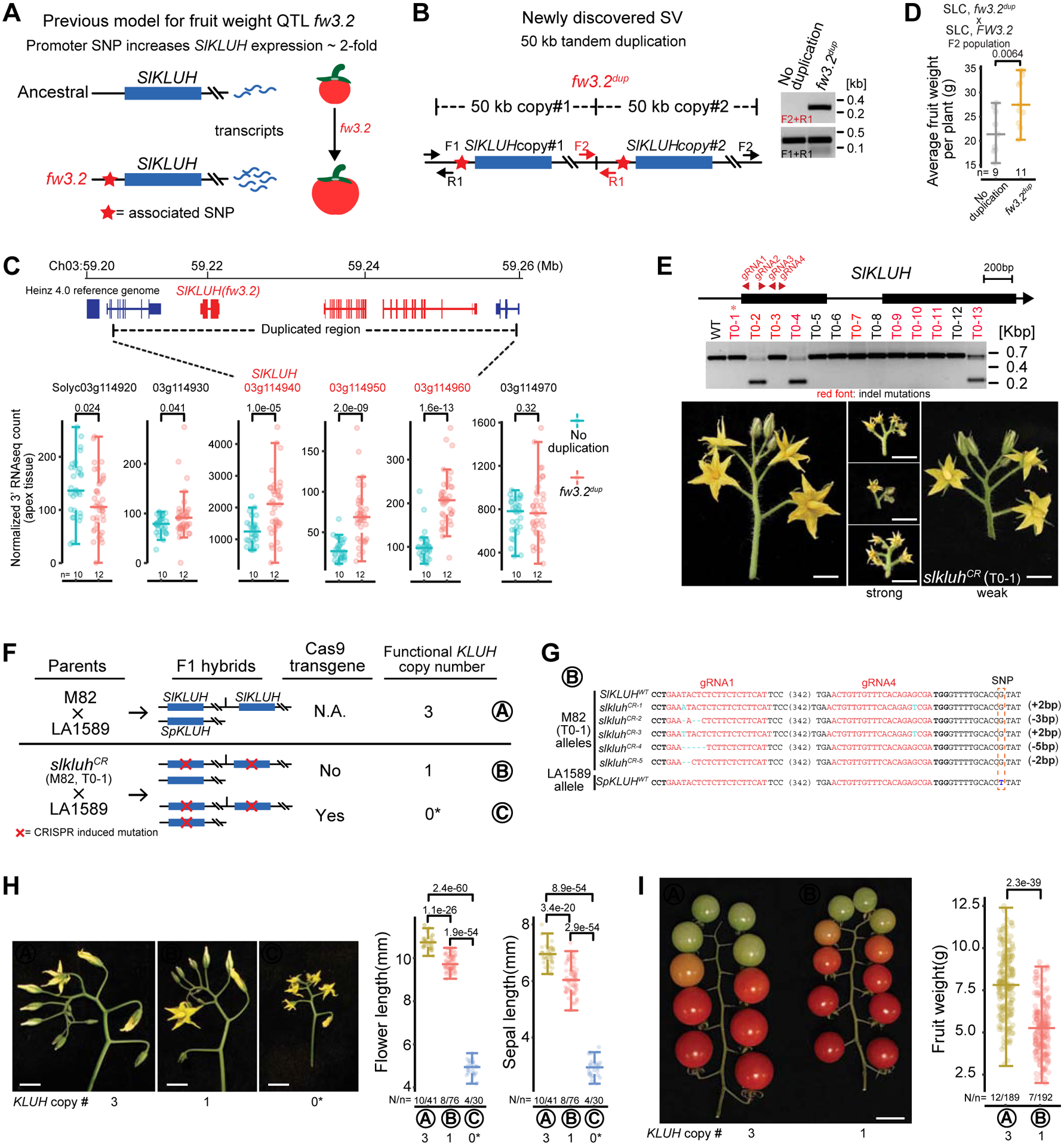
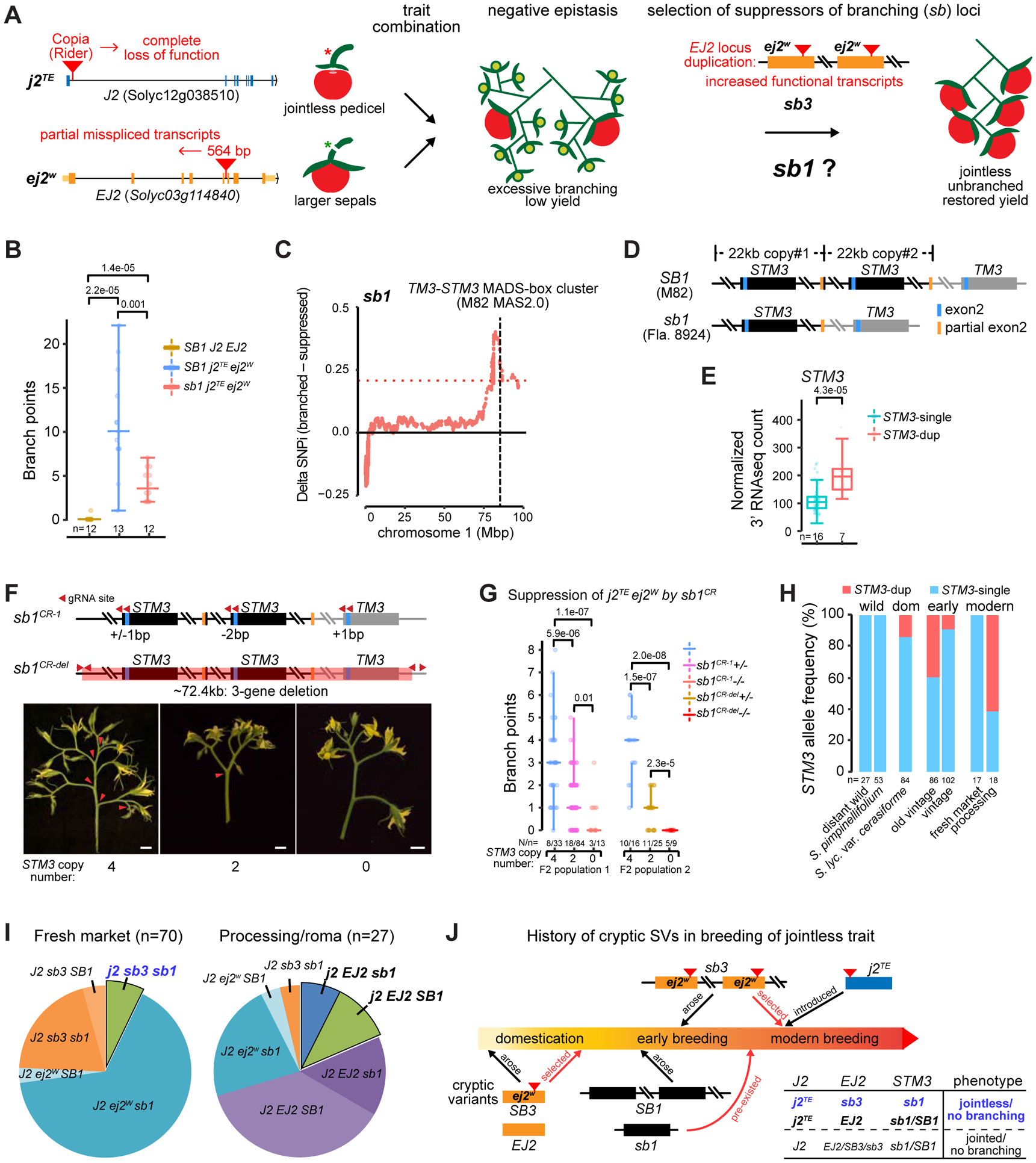
Structural variants (SVs) underlie important crop improvement and domestication traits. However, resolving the extent, diversity, and quantitative impact of SVs has been challenging. We used long-read nanopore sequencing to capture 238,490 SVs in 100 diverse tomato lines. This panSV genome, along with 14 new reference assemblies, revealed large-scale intermixing of diverse genotypes, as well as thousands of SVs intersecting genes and cis-regulatory regions. Hundreds of SV-gene pairs exhibit subtle and significant expression changes, which could broadly influence quantitative trait variation. By combining quantitative genetics with genome editing, we show how multiple SVs that changed gene dosage and expression levels modified fruit flavor, size, and production. In the last example, higher order epistasis among four SVs affecting three related transcription factors allowed introduction of an important harvesting trait in modern tomato. Our findings highlight the underexplored role of SVs in genotype-to-phenotype relationships and their widespread importance and utility in crop improvement.
Keywords: breeding; cis-regulatory; copy number variation; cryptic variation; domestication; dosage; epistasis; long-read sequencing; structural variation; tomato.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests W.R.M. is a founder and shareholder of Orion Genomics, a plant genetics company. Z.B.L. is a consultant for and a member of the Scientific Strategy Board of Inari Agriculture. Orion Genomics and Inari Agriculture had no role in the planning, execution, or analysis of the experiments described here.
Figures
Comment in
-
Resolving the roles of structural variants.Nat Rev Genet. 2020 Sep;21(9):507. doi: 10.1038/s41576-020-0264-6. Nat Rev Genet. 2020. PMID: 32636498 No abstract available.
-
Mobile Transposable Elements Shape Plant Genome Diversity.Trends Plant Sci. 2020 Nov;25(11):1062-1064. doi: 10.1016/j.tplants.2020.08.003. Epub 2020 Aug 28. Trends Plant Sci. 2020. PMID: 32863103
References
-
- Aflitos S, Schijlen E, De Jong H, De Ridder D, Smit S, Finkers R, Wang J, Zhang G, Li N, Mao L, et al. (2014). Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 80, 136–148. - PubMed
-
- Aflitos SA, Sanchez-Perez G, de Ridder D, Fransz P, Schranz ME, de Jong H, and Peters SA (2015). Introgression browser: high-throughput whole-genome SNP visualization. Plant J. 82, 174–182. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
