

The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy
- PMID: 32555305
- PMCID: PMC7300032
- DOI: 10.1038/s41598-020-66656-9
The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unreponsive to standard heart failure therapy
Erratum in
-
Author Correction: The phospholamban p.(Arg14del) pathogenic variant leads to cardiomyopathy with heart failure and is unresponsive to standard heart failure therapy.Sci Rep. 2020 Oct 2;10(1):16710. doi: 10.1038/s41598-020-70780-x. Sci Rep. 2020. PMID: 33009422 Free PMC article.
Abstract
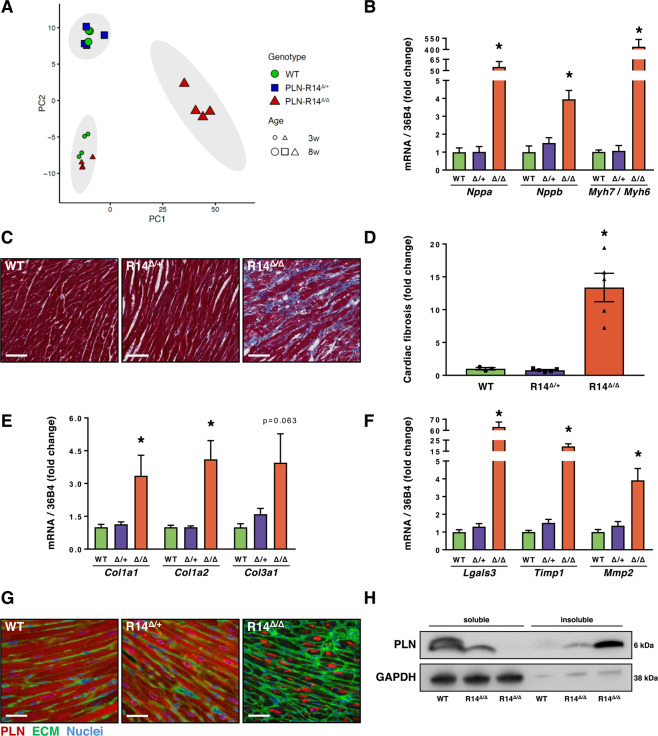
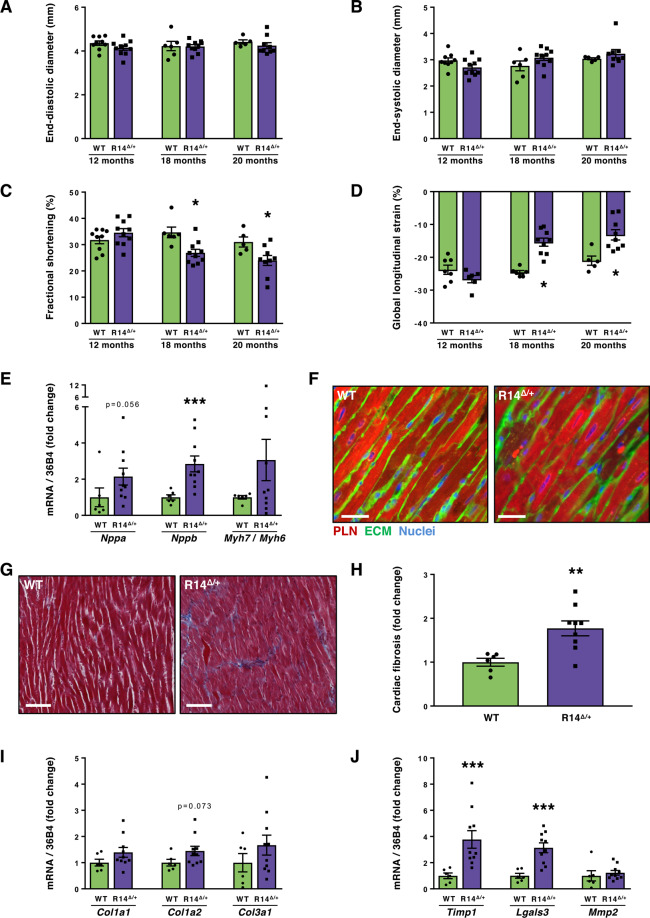
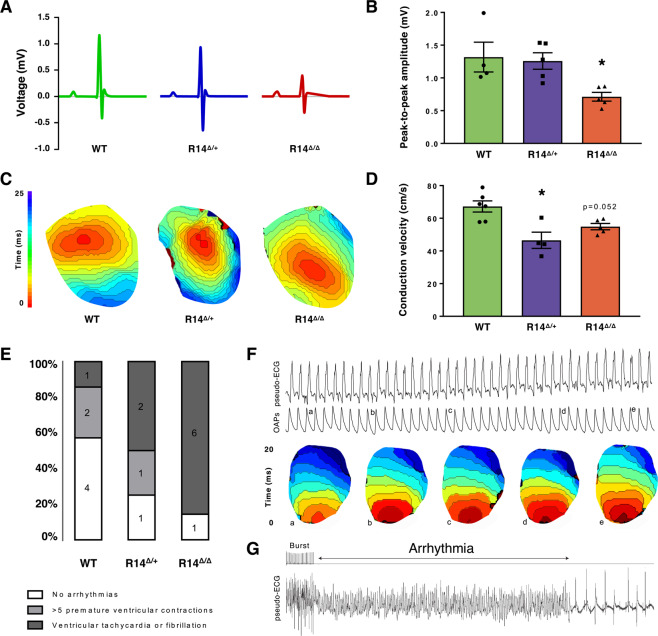
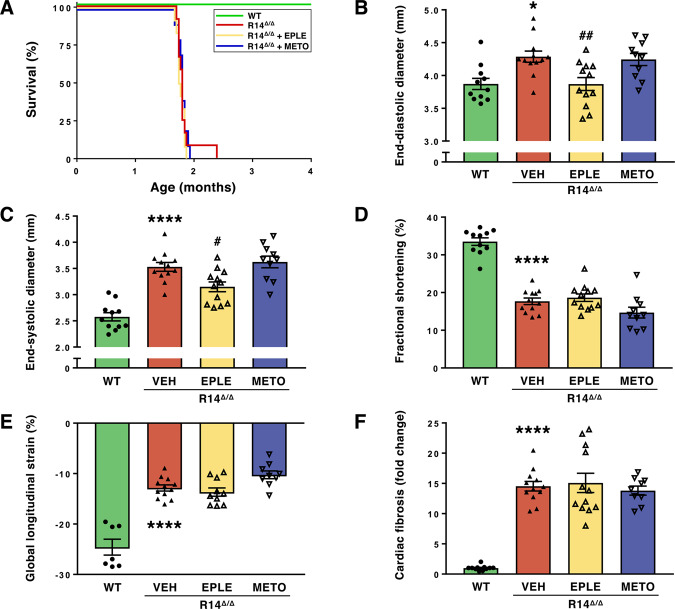
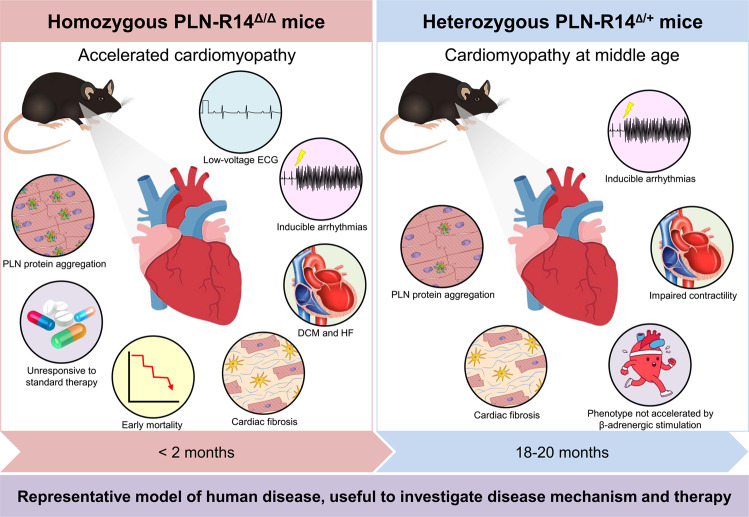
Phospholamban (PLN) plays a role in cardiomyocyte calcium handling as primary inhibitor of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA). The p.(Arg14del) pathogenic variant in the PLN gene results in a high risk of developing dilated or arrhythmogenic cardiomyopathy with heart failure. There is no established treatment other than standard heart failure therapy or heart transplantation. In this study, we generated a novel mouse model with the PLN-R14del pathogenic variant, performed detailed phenotyping, and tested the efficacy of established heart failure therapies eplerenone or metoprolol. Heterozygous PLN-R14del mice demonstrated increased susceptibility to ex vivo induced arrhythmias, and cardiomyopathy at 18 months of age, which was not accelerated by isoproterenol infusion. Homozygous PLN-R14del mice exhibited an accelerated phenotype including cardiac dilatation, contractile dysfunction, decreased ECG potentials, high susceptibility to ex vivo induced arrhythmias, myocardial fibrosis, PLN protein aggregation, and early mortality. Neither eplerenone nor metoprolol administration improved cardiac function or survival. In conclusion, our novel PLN-R14del mouse model exhibits most features of human disease. Administration of standard heart failure therapy did not rescue the phenotype, underscoring the need for better understanding of the pathophysiology of PLN-R14del-associated cardiomyopathy. This model provides a great opportunity to study the pathophysiology, and to screen for potential therapeutic treatments.
Conflict of interest statement
The University Medical Center Groningen, which employs the majority of the authors, has received research grants and/or fees from Abbott, AstraZeneca, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche. R.A.d.B. is a minority shareholder of scPharmaceuticals, and received personal fees from Abbott, AstraZeneca, MandalMed. and Novartis. The other authors have no competing interests.
Figures

References
-
- MacLennan DH, Kranias EG. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003;4:566–577. - PubMed
-
- DeWitt MM, MacLeod HM, Soliven B, McNally EM. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. J. Am. Coll. Cardiol. 2006;48:1396–1398. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
