

Determination of RNA structural diversity and its role in HIV-1 RNA splicing
- PMID: 32555469
- PMCID: PMC7310298
- DOI: 10.1038/s41586-020-2253-5
Determination of RNA structural diversity and its role in HIV-1 RNA splicing
Erratum in
-
Author Correction: Determination of RNA structural diversity and its role in HIV-1 RNA splicing.Nature. 2020 Dec;588(7837):E16. doi: 10.1038/s41586-020-2949-6. Nature. 2020. PMID: 33214714
Abstract
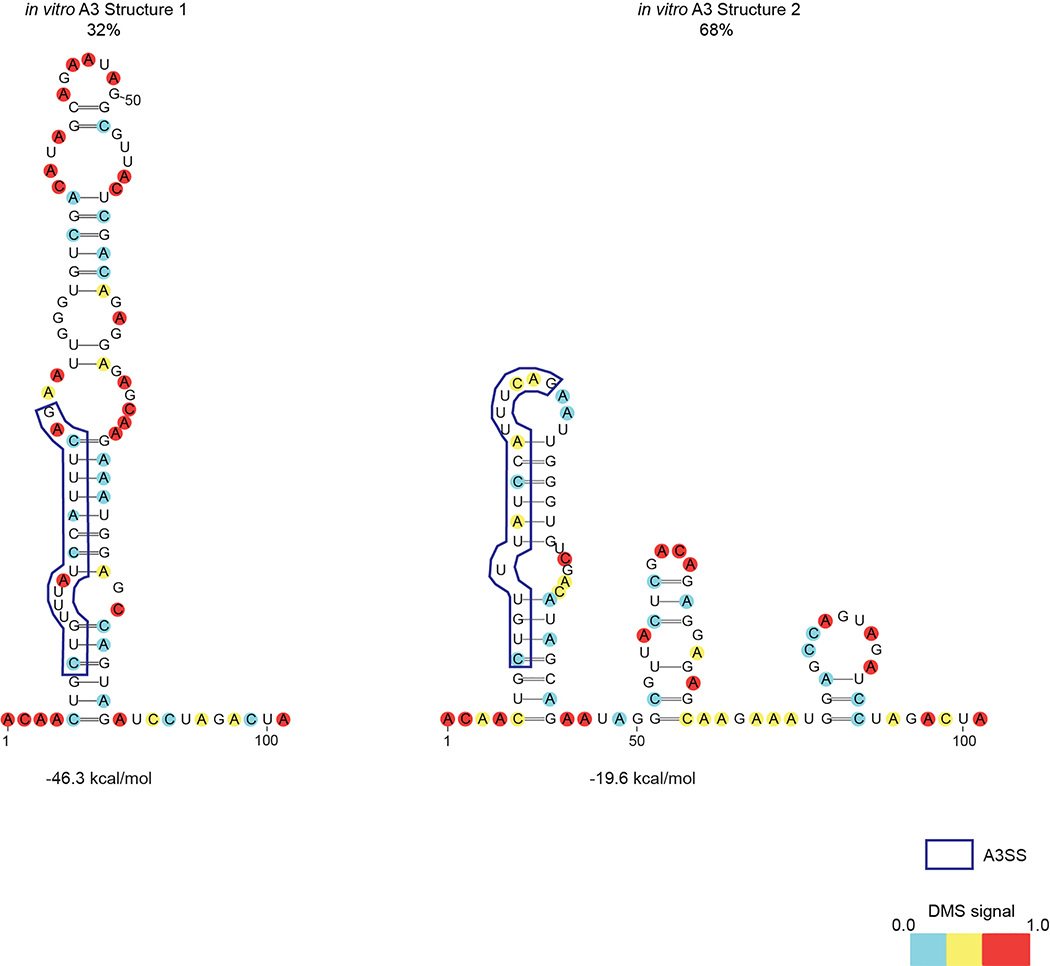
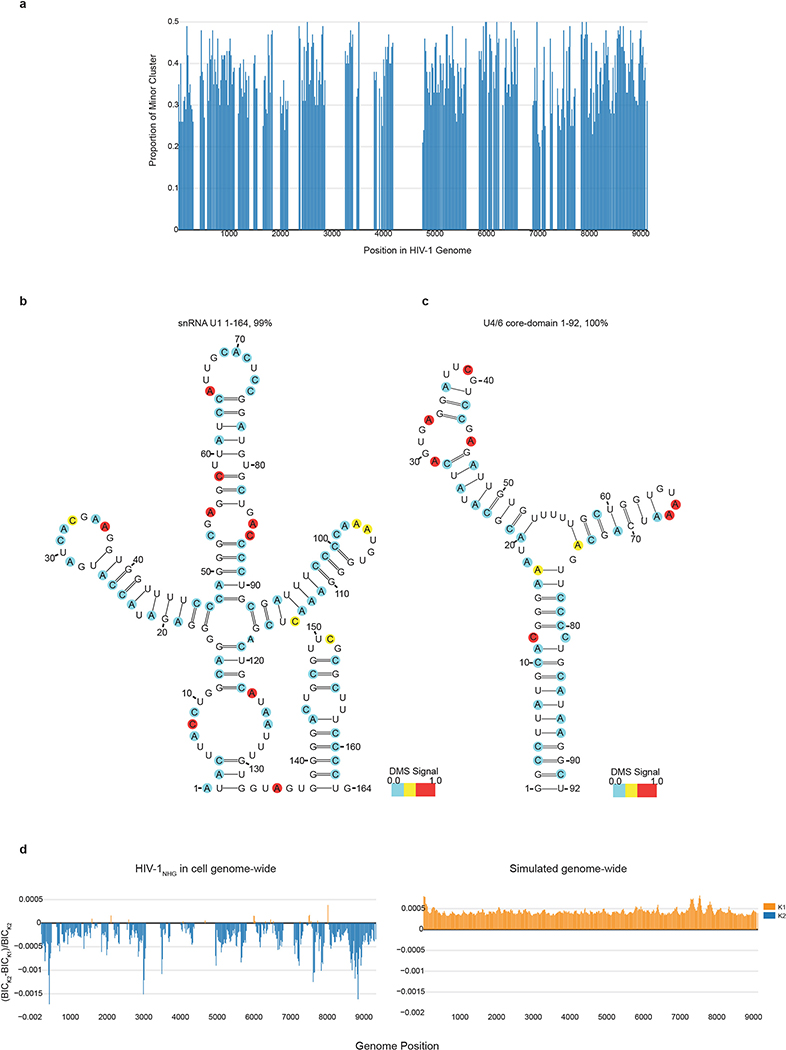
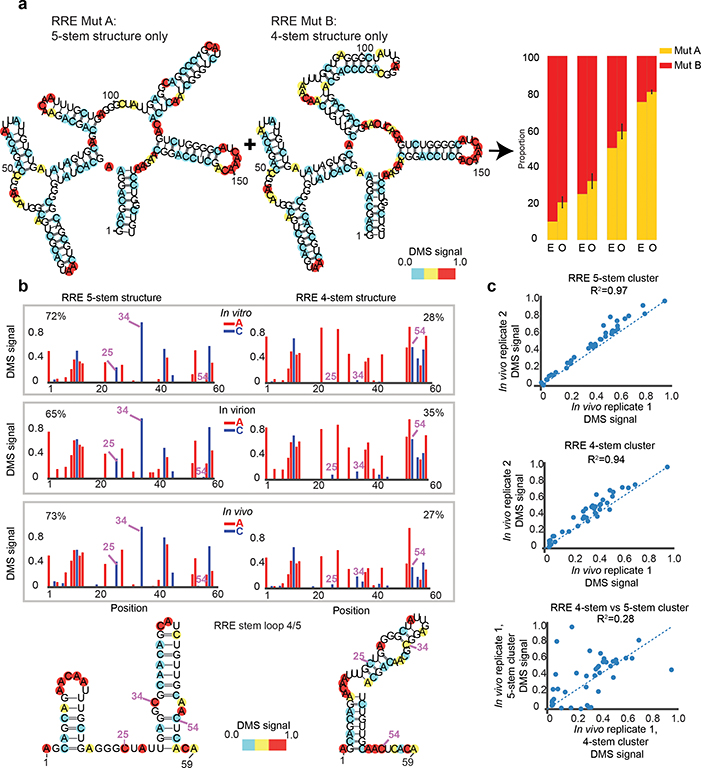
Human immunodeficiency virus 1 (HIV-1) is a retrovirus with a ten-kilobase single-stranded RNA genome. HIV-1 must express all of its gene products from a single primary transcript, which undergoes alternative splicing to produce diverse protein products that include structural proteins and regulatory factors1,2. Despite the critical role of alternative splicing, the mechanisms that drive the choice of splice site are poorly understood. Synonymous RNA mutations that lead to severe defects in splicing and viral replication indicate the presence of unknown cis-regulatory elements3. Here we use dimethyl sulfate mutational profiling with sequencing (DMS-MaPseq) to investigate the structure of HIV-1 RNA in cells, and develop an algorithm that we name 'detection of RNA folding ensembles using expectation-maximization' (DREEM), which reveals the alternative conformations that are assumed by the same RNA sequence. Contrary to previous models that have analysed population averages4, our results reveal heterogeneous regions of RNA structure across the entire HIV-1 genome. In addition to confirming that in vitro characterized5 alternative structures for the HIV-1 Rev responsive element also exist in cells, we discover alternative conformations at critical splice sites that influence the ratio of transcript isoforms. Our simultaneous measurement of splicing and intracellular RNA structure provides evidence for the long-standing hypothesis6-8 that heterogeneity in RNA conformation regulates splice-site use and viral gene expression.
Conflict of interest statement
[Ethics declarations]
The authors declare no competing interests.
Figures












References
[References]
[Methods References]
-
- Lahm HW & Stein S Characterization of recombinant human IL-2 with micromethods. Journal of Chromatography 326, 357–361 (1985). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
