

Comprehensive genetic analysis of 961 unrelated Duchenne Muscular Dystrophy patients: Focus on diagnosis, prevention and therapeutic possibilities
- PMID: 32559196
- PMCID: PMC7304910
- DOI: 10.1371/journal.pone.0232654
Comprehensive genetic analysis of 961 unrelated Duchenne Muscular Dystrophy patients: Focus on diagnosis, prevention and therapeutic possibilities
Abstract
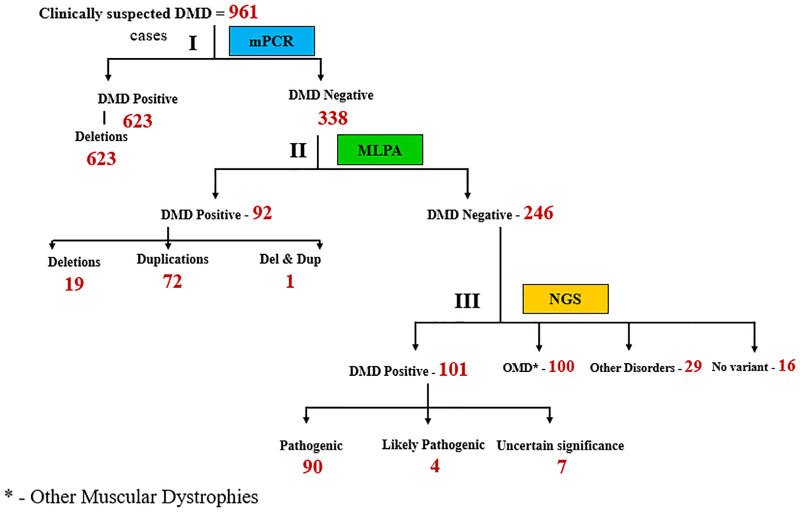
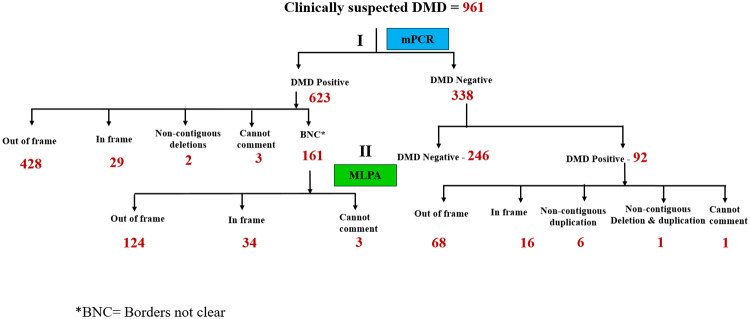
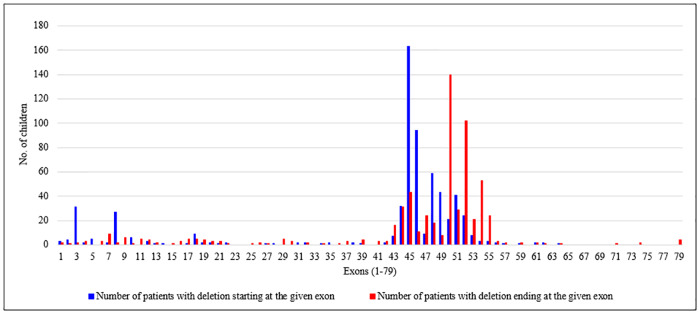
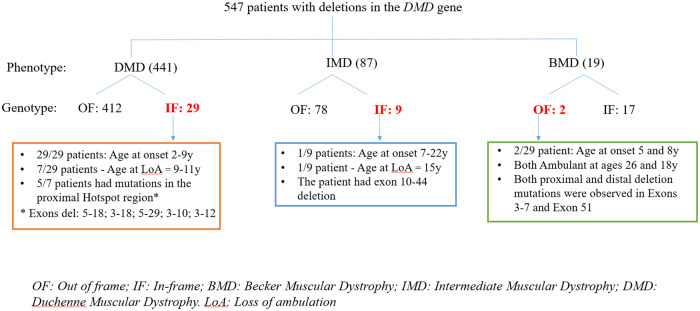
Recently DNA sequencing analysis has played a vital role in the unambiguous diagnosis of clinically suspected patients with Duchenne Muscular Dystrophy (DMD). DMD is a monogenic, X-linked, recessive, degenerative pediatric neuromuscular disorder affecting males, invariably leading to fatal cardiopulmonary failure. Early and precise diagnosis of the disease is an essential part of an effective disease management strategy as care guidelines and prevention through counseling need to be initiated at the earliest particularly since therapies are now available for a subset of patients. In this manuscript we report the DMD gene mutational profiles of 961 clinically suspected male DMD patients, 99% of whom were unrelated. We utilized a molecular diagnostic approach which is cost-effective for most patients and follows a systematic process that sequentially involves identification of hotspot deletions using mPCR, large deletions and duplications using MLPA and small insertions/ deletions and point mutations using an NGS muscular dystrophy gene panel. Pathogenic DMD gene mutations were identified in 84% of patients. Our data compared well with the frequencies and distribution of deletions and duplications reported in the DMD gene in other published studies. We also describe a number of rare in-frame mutations, which appeared to be enriched in the 5' proximal hotspot region of the DMD gene. Furthermore, we identified a family with a rare non-contiguous deletion mutation in the DMD gene where three males were affected and two females were deemed carriers. A subset of patients with mutations in the DMD gene who are likely to benefit therapeutically from new FDA and EMA approved drugs were found in our cohort. Given that the burden of care for DMD patients invariably falls on the mothers, particularly in rural India, effective genetic counseling followed by carrier screening is crucial for prevention of this disorder. We analyzed the carrier status of consented female relatives of 463 probands to gauge the percentage of patients with familial disease. Our analysis revealed 43.7% of mothers with DMD gene mutations. Our comprehensive efforts, involving complete genetic testing coupled with compassionate genetic counseling provided to DMD patients and their families, are intended to improve the quality of life of DMD patients and to empower carrier females to make informed reproductive choices to impede the propagation of this deadly disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
