

Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress
- PMID: 32559414
- PMCID: PMC7415721
- DOI: 10.1016/j.cmet.2020.06.002
Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress
Abstract
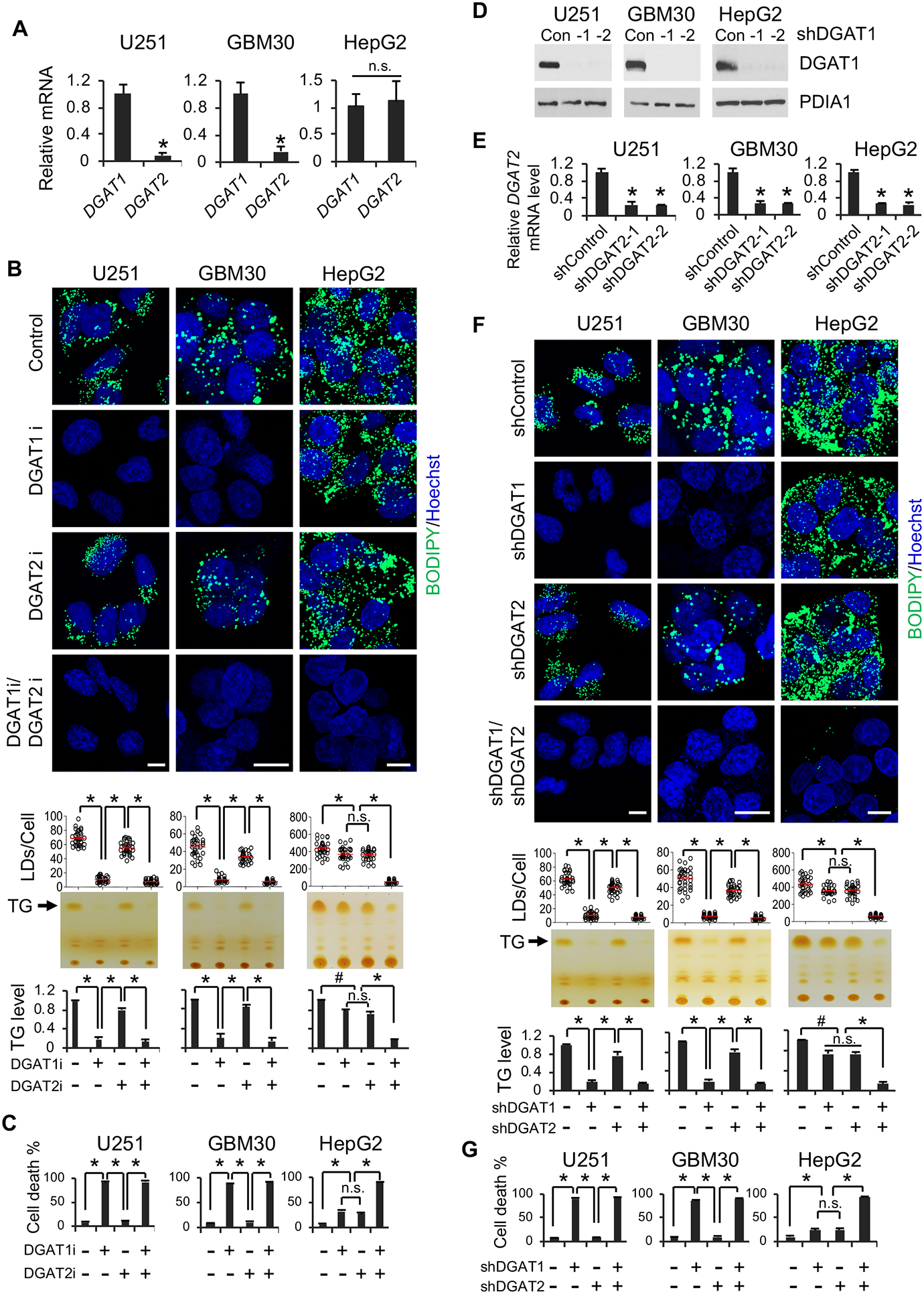
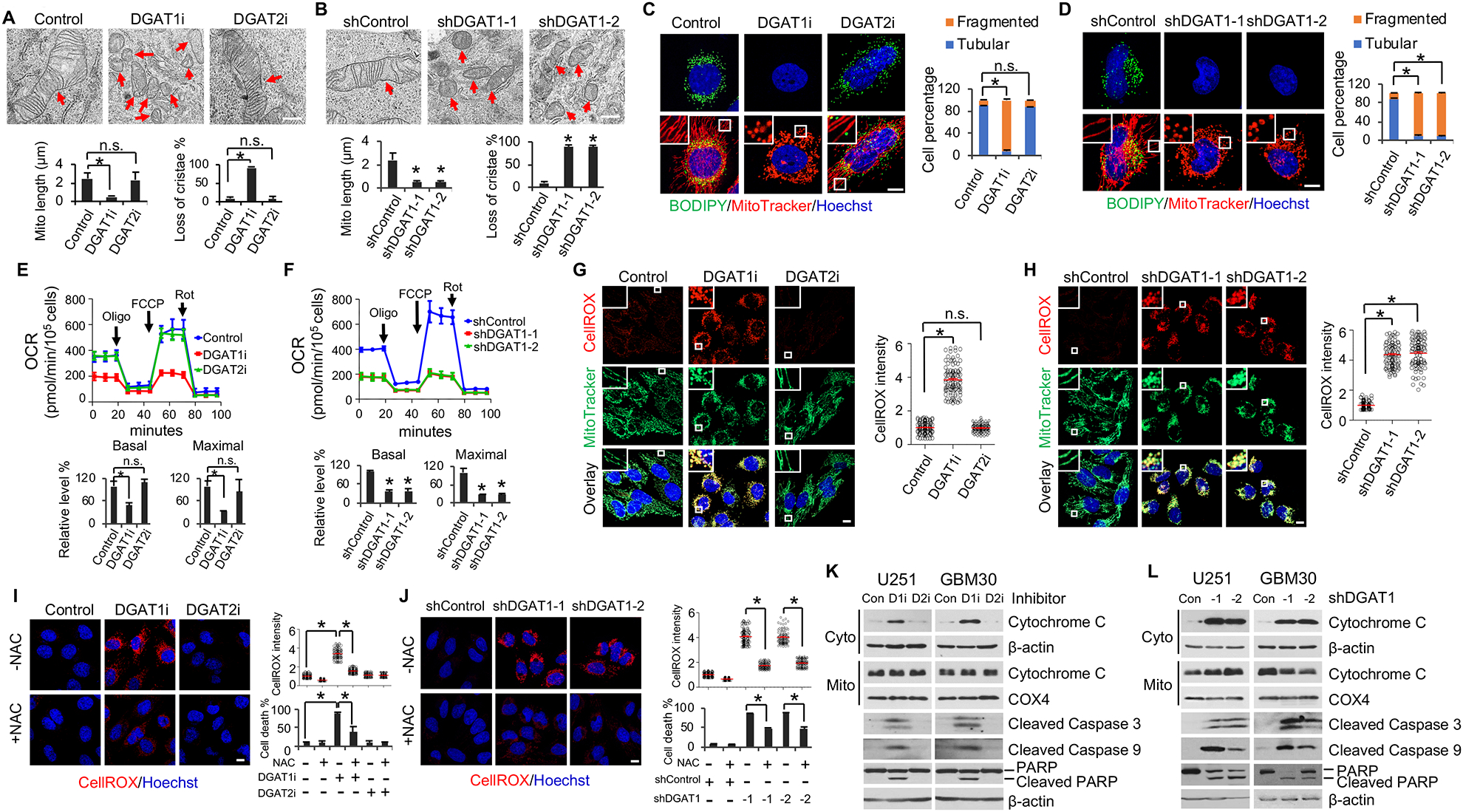
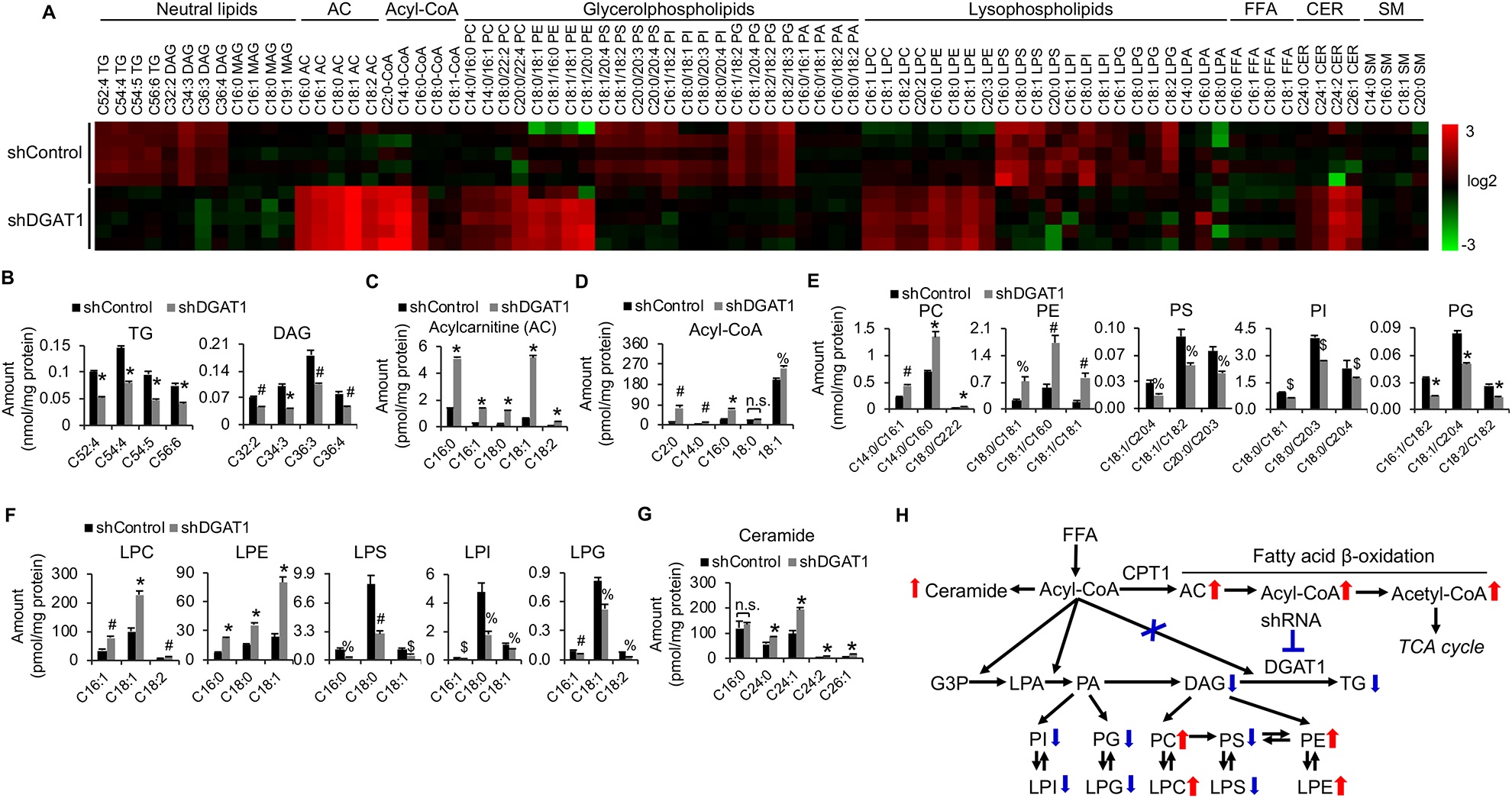
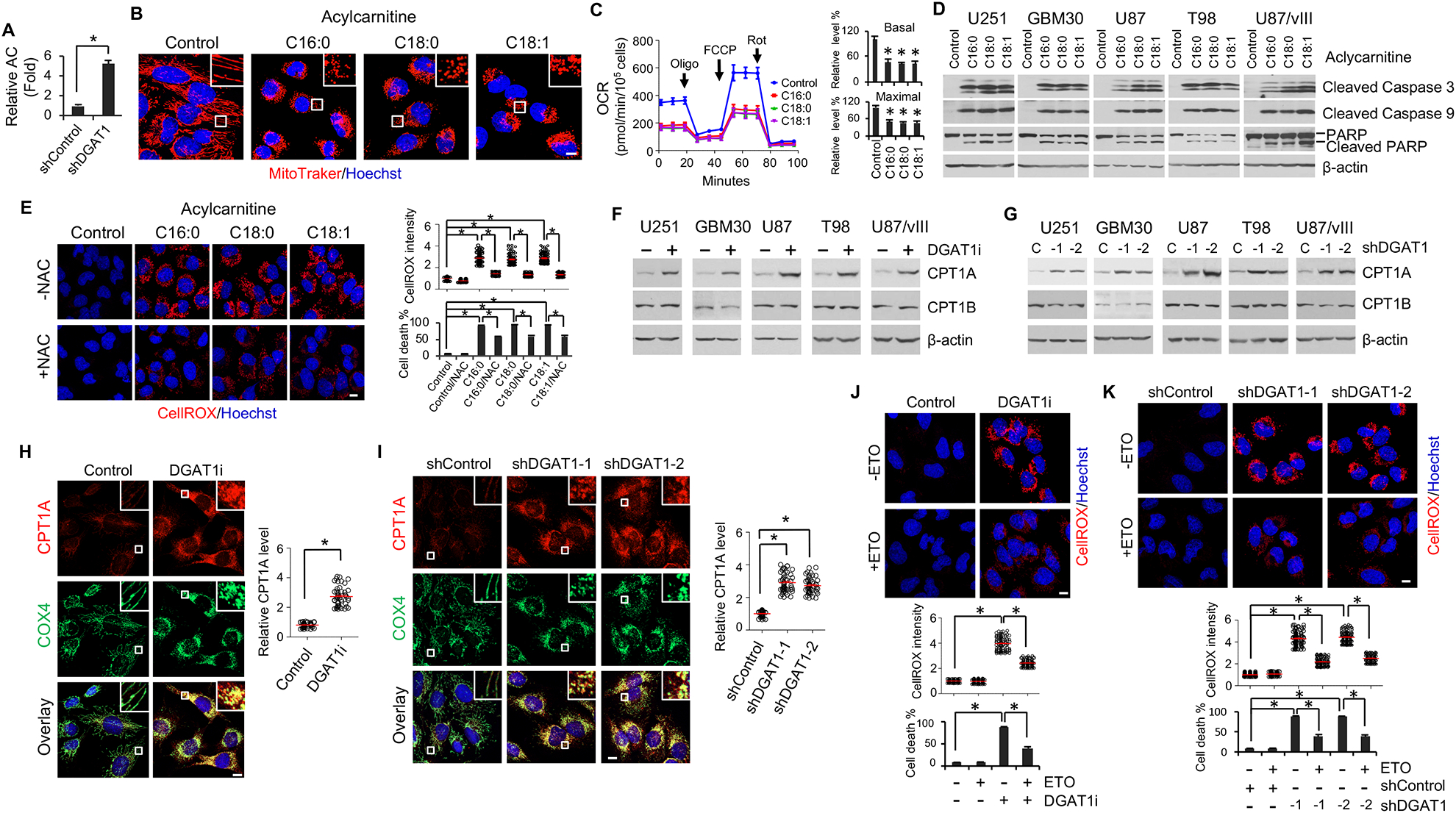
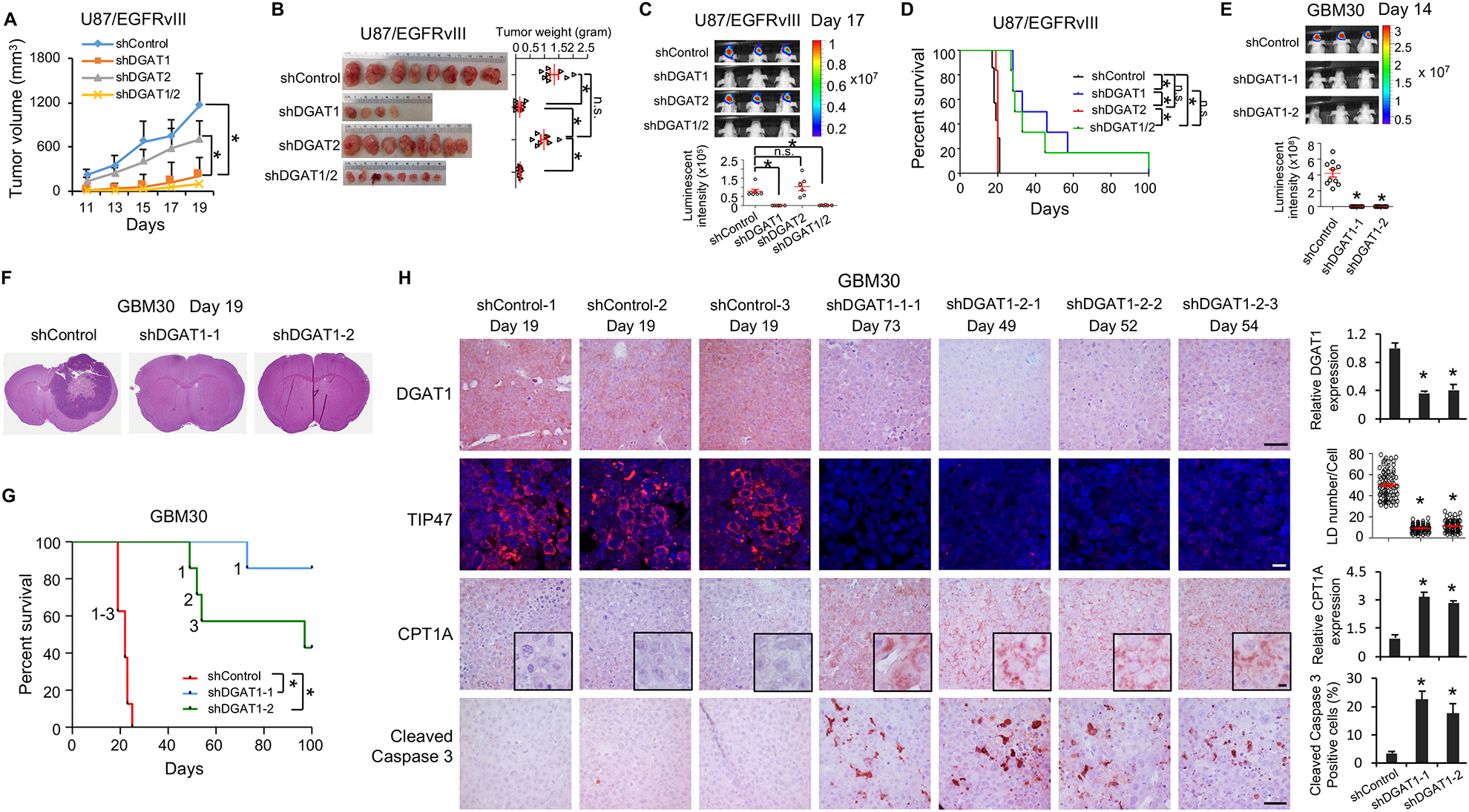
Glioblastoma (GBM), a mostly lethal brain tumor, acquires large amounts of free fatty acids (FAs) to promote cell growth. But how the cancer avoids lipotoxicity is unknown. Here, we identify that GBM upregulates diacylglycerol-acyltransferase 1 (DGAT1) to store excess FAs into triglycerides and lipid droplets. Inhibiting DGAT1 disrupted lipid homeostasis and resulted in excessive FAs moving into mitochondria for oxidation, leading to the generation of high levels of reactive oxygen species (ROS), mitochondrial damage, cytochrome c release, and apoptosis. Adding N-acetyl-cysteine or inhibiting FA shuttling into mitochondria decreased ROS and cell death induced by DGAT1 inhibition. We show in xenograft models that targeting DGAT1 blocked lipid droplet formation, induced tumor cell apoptosis, and markedly suppressed GBM growth. Together, our study demonstrates that DGAT1 upregulation protects GBM from oxidative damage and maintains lipid homeostasis by facilitating storage of excess FAs. Targeting DGAT1 could be a promising therapeutic approach for GBM.
Keywords: DGAT1; ROS; acylcarnitine; fatty acids; glioblastoma; lipid droplets; lipotoxicity; mitochondria; oxidative stress; triglycerides.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare that there are no competing interests.
Figures


References
-
- Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, and Viola JP (2008). Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 68, 1732–1740. - PubMed
-
- Aruoma OI, Halliwell B, Hoey BM, and Butler J (1989). The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 6, 593–597. - PubMed
-
- Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, Stanley TB, Sanders B, Goetz A, Gaul N, et al. (2013). Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res 73, 1993–2002. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
