

Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids
- PMID: 32559461
- PMCID: PMC7339148
- DOI: 10.1016/j.cell.2020.05.053
Metabolic Fingerprinting Links Oncogenic PIK3CA with Enhanced Arachidonic Acid-Derived Eicosanoids
Abstract
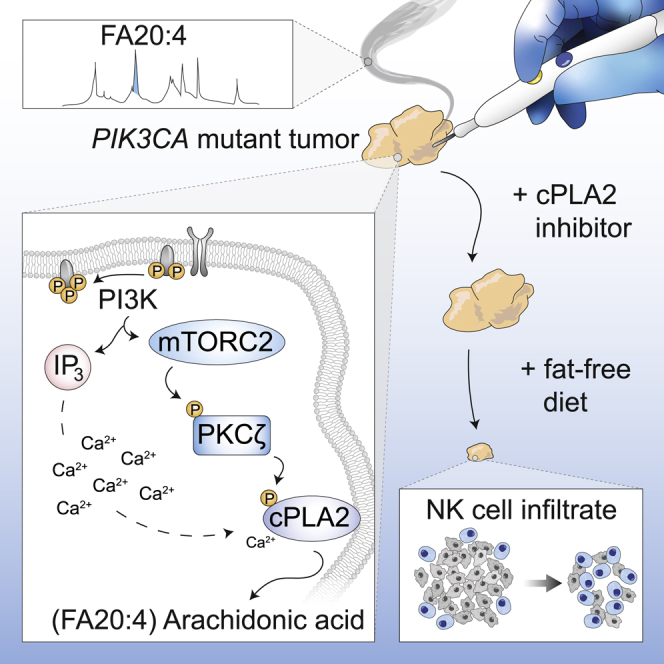
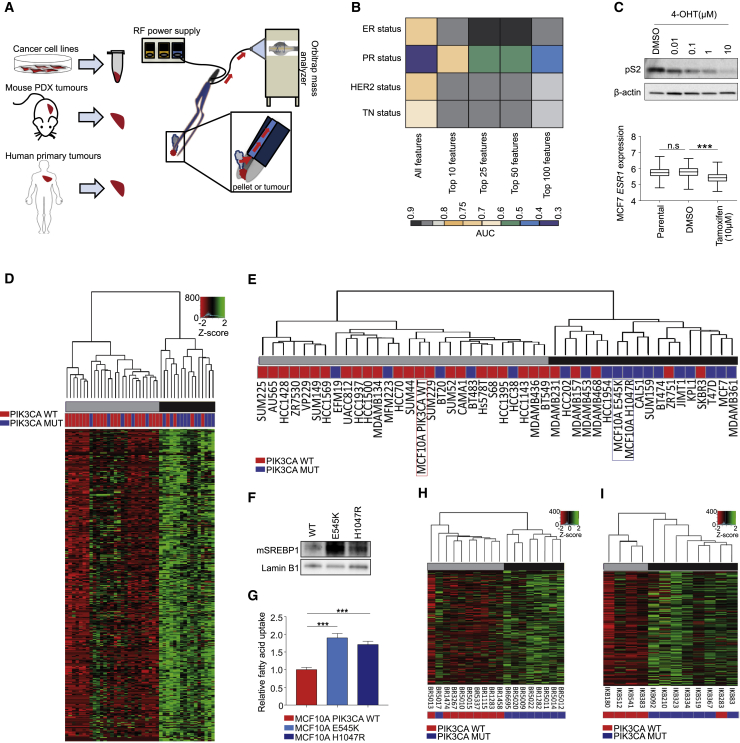
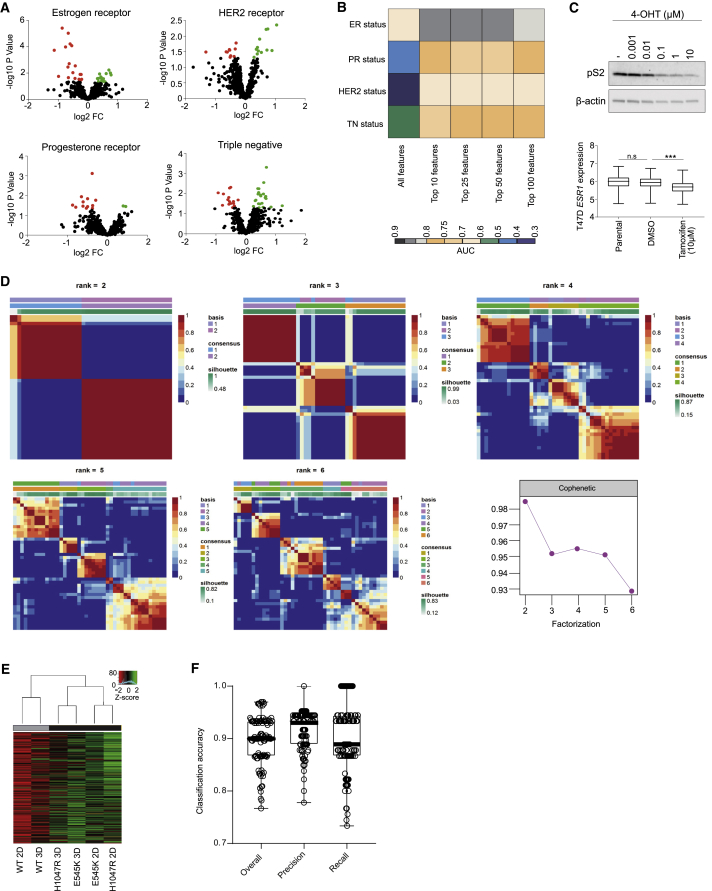
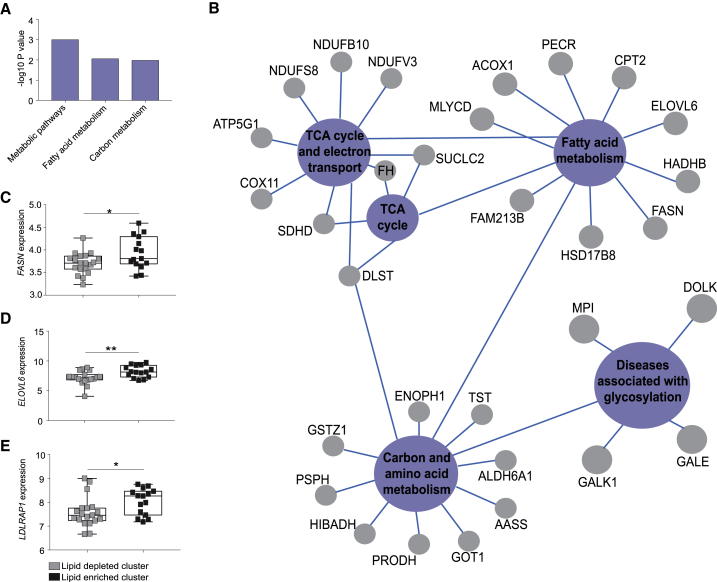
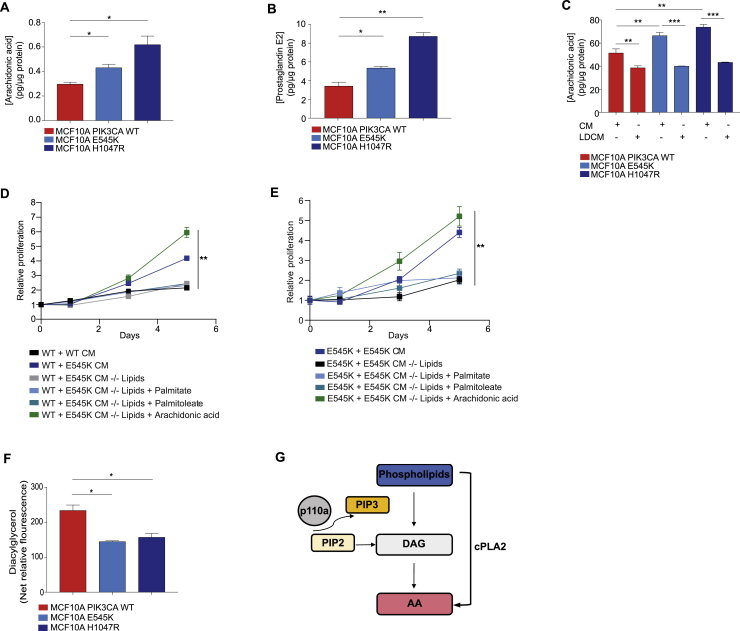
Oncogenic transformation is associated with profound changes in cellular metabolism, but whether tracking these can improve disease stratification or influence therapy decision-making is largely unknown. Using the iKnife to sample the aerosol of cauterized specimens, we demonstrate a new mode of real-time diagnosis, coupling metabolic phenotype to mutant PIK3CA genotype. Oncogenic PIK3CA results in an increase in arachidonic acid and a concomitant overproduction of eicosanoids, acting to promote cell proliferation beyond a cell-autonomous manner. Mechanistically, mutant PIK3CA drives a multimodal signaling network involving mTORC2-PKCζ-mediated activation of the calcium-dependent phospholipase A2 (cPLA2). Notably, inhibiting cPLA2 synergizes with fatty acid-free diet to restore immunogenicity and selectively reduce mutant PIK3CA-induced tumorigenicity. Besides highlighting the potential for metabolic phenotyping in stratified medicine, this study reveals an important role for activated PI3K signaling in regulating arachidonic acid metabolism, uncovering a targetable metabolic vulnerability that largely depends on dietary fat restriction. VIDEO ABSTRACT.
Keywords: PIK3CA; PKCζ; arachidonic acid; cPLA2; cancer metabolism; diet; eicosanoids; fat restriction; iKnife; mTORC2.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests N.K. and G.P. are inventors on a patent application covering new methods and compositions useful in the treatment of cancers with PIK3CA mutation (application number GB 2005874.9).
Figures
Comment in
-
Metabolites going up in smoke.Nat Rev Cancer. 2020 Sep;20(9):482. doi: 10.1038/s41568-020-0289-3. Nat Rev Cancer. 2020. PMID: 32636490 No abstract available.
-
On the Cutting Edge of Translational Research.Clin Chem. 2021 Mar 31;67(4):583-584. doi: 10.1093/clinchem/hvaa273. Clin Chem. 2021. PMID: 33230521 Free PMC article. No abstract available.
References
-
- Alexander L.D., Ding Y., Alagarsamy S., Cui X.L., Douglas J.G. Arachidonic acid induces ERK activation via Src SH2 domain association with the epidermal growth factor receptor. Kidney Int. 2006;69:1823–1832. - PubMed
-
- Alexander J., Gildea L., Balog J., Speller A., McKenzie J., Muirhead L., Scott A., Kontovounisios C., Rasheed S., Teare J. A novel methodology for in vivo endoscopic phenotyping of colorectal cancer based on real-time analysis of the mucosal lipidome: a prospective observational study of the iKnife. Surg. Endosc. 2017;31:1361–1370. - PMC - PubMed
-
- Balog J., Sasi-Szabó L., Kinross J., Lewis M.R., Muirhead L.J., Veselkov K., Mirnezami R., Dezső B., Damjanovich L., Darzi A. Intraoperative tissue identification using rapid evaporative ionization mass spectrometry. Sci. Transl. Med. 2013;5:194ra93. - PubMed
-
- Basingab F.S., Ahmadi M., Morgan D.J. IFNγ-Dependent Interactions between ICAM-1 and LFA-1 Counteract Prostaglandin E2-Mediated Inhibition of Antitumor CTL Responses. Cancer Immunol. Res. 2016;4:400–411. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
