

Hybrid Gene Origination Creates Human-Virus Chimeric Proteins during Infection
- PMID: 32559462
- PMCID: PMC7323901
- DOI: 10.1016/j.cell.2020.05.035
Hybrid Gene Origination Creates Human-Virus Chimeric Proteins during Infection
Abstract
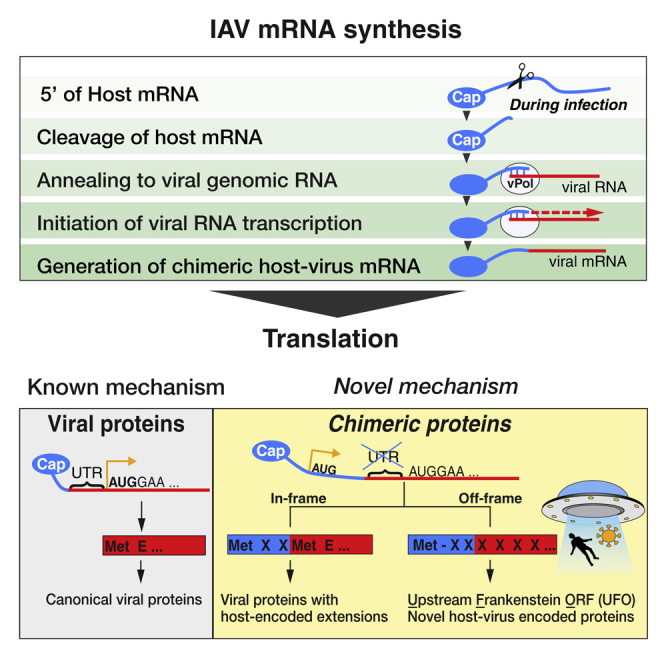
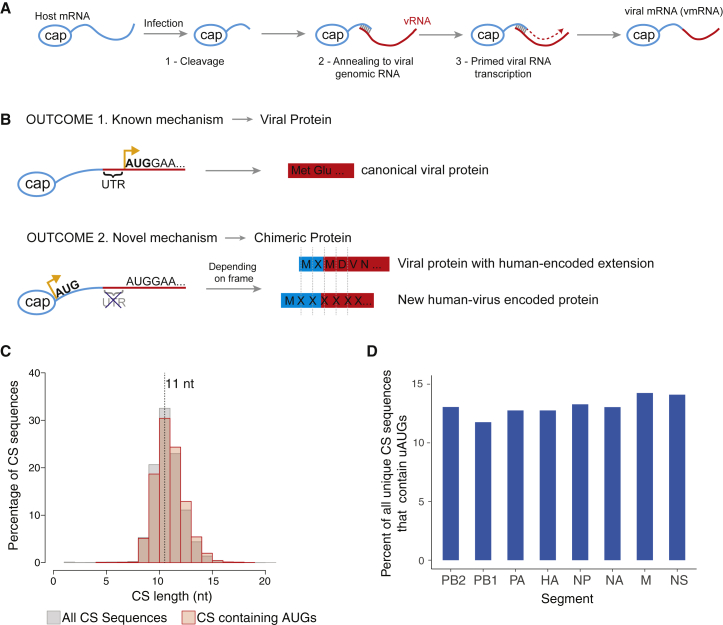
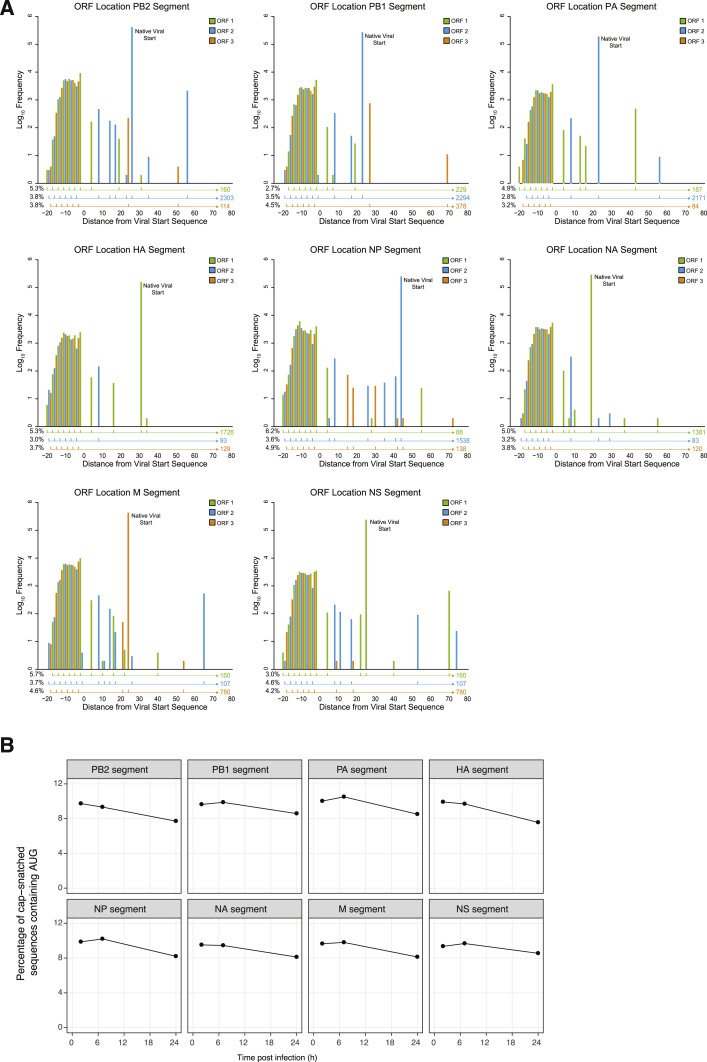
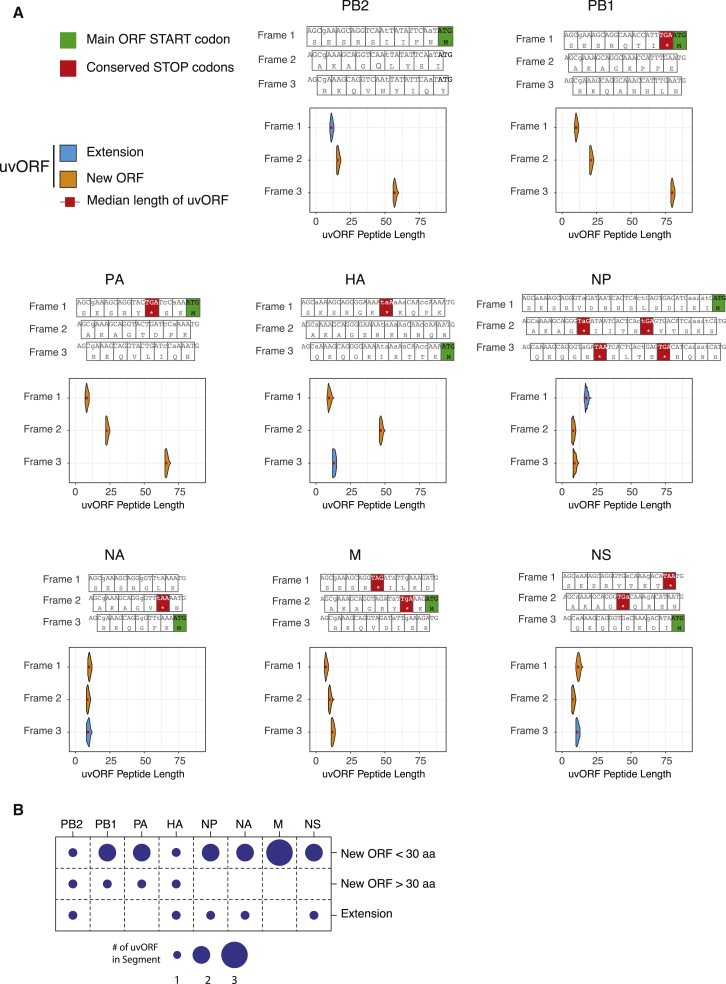
RNA viruses are a major human health threat. The life cycles of many highly pathogenic RNA viruses like influenza A virus (IAV) and Lassa virus depends on host mRNA, because viral polymerases cleave 5'-m7G-capped host transcripts to prime viral mRNA synthesis ("cap-snatching"). We hypothesized that start codons within cap-snatched host transcripts could generate chimeric human-viral mRNAs with coding potential. We report the existence of this mechanism of gene origination, which we named "start-snatching." Depending on the reading frame, start-snatching allows the translation of host and viral "untranslated regions" (UTRs) to create N-terminally extended viral proteins or entirely novel polypeptides by genetic overprinting. We show that both types of chimeric proteins are made in IAV-infected cells, generate T cell responses, and contribute to virulence. Our results indicate that during infection with IAV, and likely a multitude of other human, animal and plant viruses, a host-dependent mechanism allows the genesis of hybrid genes.
Keywords: RNA hybrid; cap-snatching; chimeric proteins; gene origination; influenza; segmented negative-strand RNA viruses; uORFs; upstream AUG; viral RNA; viral evolution.
Copyright © 2020 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures

Comment in
-
Cap-Snatching Leads to Novel Viral Proteins.Cell. 2020 Jun 25;181(7):1450-1451. doi: 10.1016/j.cell.2020.05.044. Cell. 2020. PMID: 32589954
-
Human-viral chimera: a novel protein affecting viral virulence and driving host T-cell immunity.Signal Transduct Target Ther. 2020 Aug 25;5(1):167. doi: 10.1038/s41392-020-00272-x. Signal Transduct Target Ther. 2020. PMID: 32843627 Free PMC article. No abstract available.
References
-
- Andrews S.J., Rothnagel J.A. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014;15:193–204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 646891/ERC_/European Research Council/International
- MR/N02995X/1/MRC_/Medical Research Council/United Kingdom
- MR/K000241/1/MRC_/Medical Research Council/United Kingdom
- 103258/Z/13/Z/WT_/Wellcome Trust/United Kingdom
- BB/J004324/1 /BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- U19 AI135972/AI/NIAID NIH HHS/United States
- MR/M011747/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12014/9/MRC_/Medical Research Council/United Kingdom
- BB/P013740/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- R01 AI113186/AI/NIAID NIH HHS/United States
- R01 AI143840/AI/NIAID NIH HHS/United States
- MR/R009945/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_12014/12/MRC_/Medical Research Council/United Kingdom
- MR/N008618/1/MRC_/Medical Research Council/United Kingdom
- 106207/WT_/Wellcome Trust/United Kingdom
- 099220/Z/12/Z/WT_/Wellcome Trust/United Kingdom
- 210703/Z/18/Z/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
