

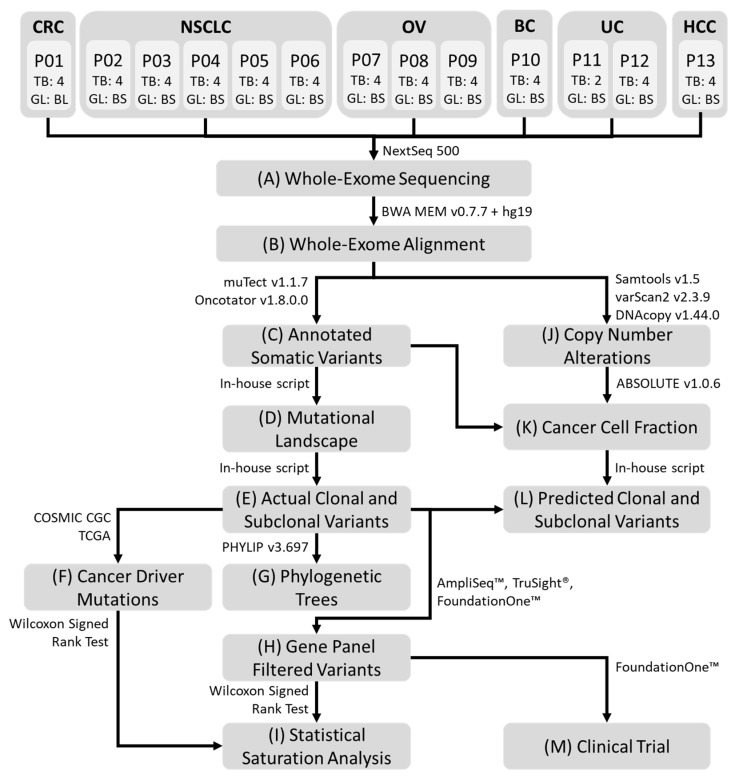
Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique
- PMID: 32560395
- PMCID: PMC7353029
- DOI: 10.3390/cancers12061599
Whole Exome Sequencing of Multi-Regional Biopsies from Metastatic Lesions to Evaluate Actionable Truncal Mutations Using a Single-Pass Percutaneous Technique
Abstract
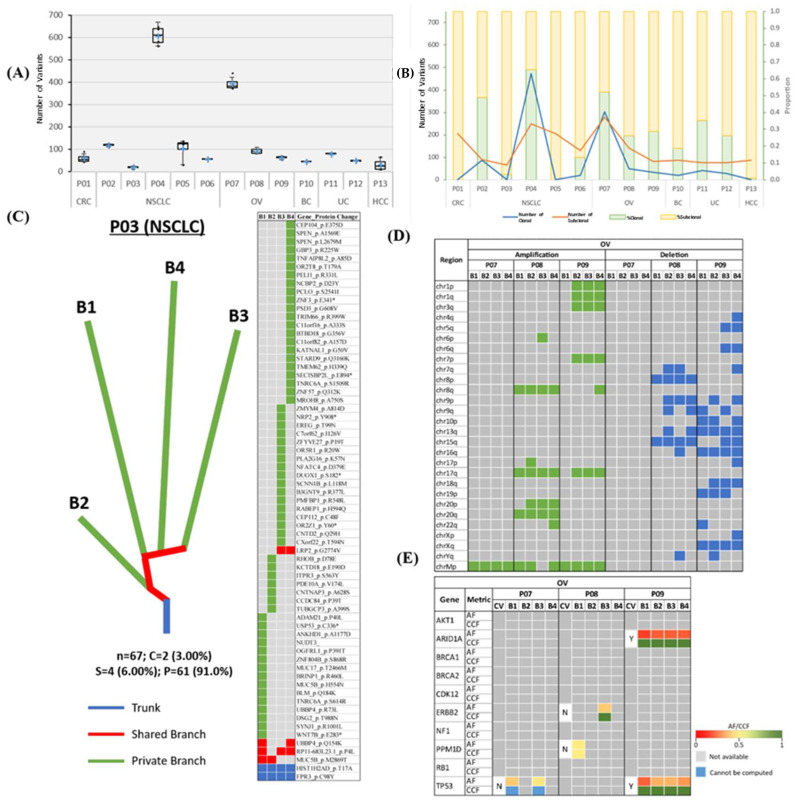
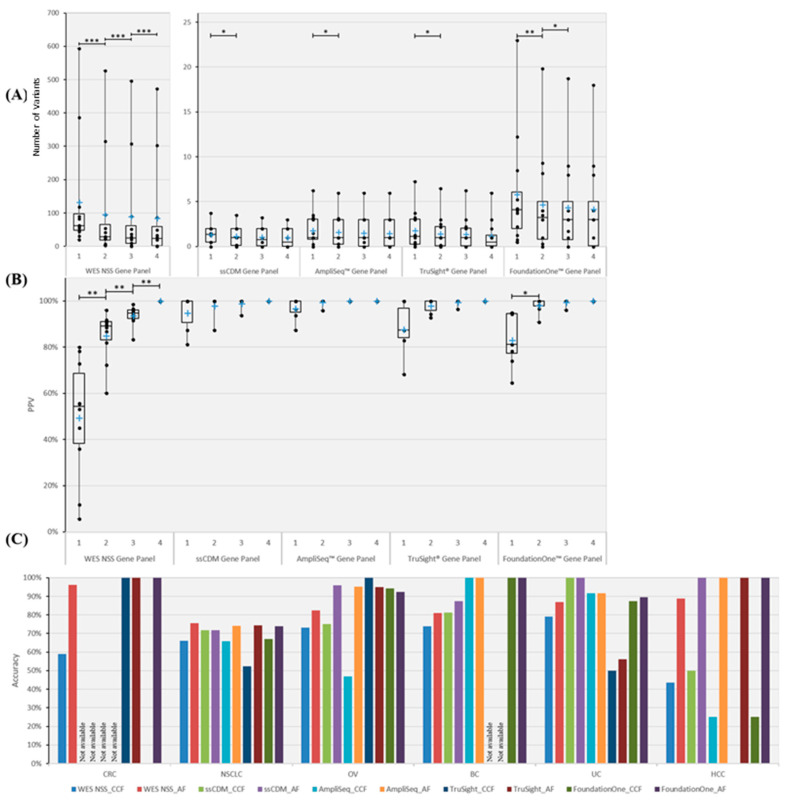
We investigate the feasibility of obtaining multiple spatially-separated biopsies from a single lesion to explore intratumor heterogeneity and identify actionable truncal mutations using whole exome sequencing (WES). A single-pass radiologically-guided percutaneous technique was used to obtain four spatially-separated biopsies from a single metastatic lesion. WES was performed to identify putative truncal variants (PTVs), defined as a non-synonymous somatic (NSS) variant present in all four spatially separated biopsies. Actionable truncal mutations-filtered using the FoundationOne panel-were defined as clinically relevant PTVs. Mutational landscapes of each biopsy and their association with patient outcomes were assessed. WES on 50 biopsied samples from 13 patients across six cancer types were analyzed. Actionable truncal mutations were identified in 9/13 patients; 31.1 ± 5.12 more unique NSS variants were detected with every additional multi- region tumor biopsy (MRTB) analyzed. The number of PTVs dropped by 16.1 ± 17.9 with every additional MRTB, with the decrease most pronounced (36.8 ± 19.7) when two MRTB were analyzed compared to one. MRTB most reliably predicted PTV compared to in silico analysis of allele frequencies and cancer cell fraction based on one biopsy sample. Three patients treated with actionable truncal mutation-directed therapy derived clinical benefit. Multi-regional sampling for genomics analysis is feasible and informative to help prioritize precision-therapy strategies.
Keywords: clonality classification; intratumor heterogeneity; multiple biopsies; strategic therapeutic intervention; tumor evolution.
Conflict of interest statement
D. Tan consultancy fees from Astra Zeneca, Roche, MSD, Merck Serono, Tessa Therapeutics, Eisai and Genmab. Reasearch funding from AstraZeneca, Bayer and Karyopharm and V. Heong consult on the advisory board for Astra Zeneca and Pfizer; the sponsor had no involvement in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review, or approval of the manuscript; or the decision to submit the manuscript for publication.
Figures
References
-
- Greaves M. Evolutionary determinants of cancer. Cancer Discov. 2015;5:806–820. doi: 10.1158/2159-8290.CD-15-0439. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
