

PARP inhibitor resistance: the underlying mechanisms and clinical implications
- PMID: 32563252
- PMCID: PMC7305609
- DOI: 10.1186/s12943-020-01227-0
PARP inhibitor resistance: the underlying mechanisms and clinical implications
Abstract
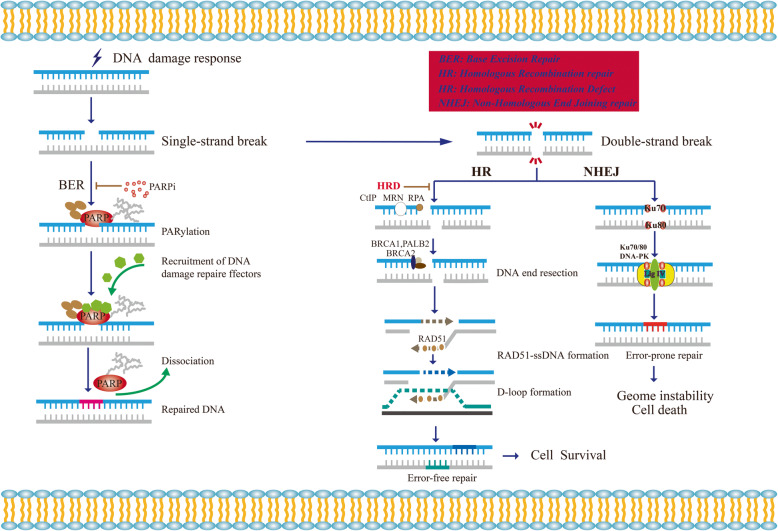
Due to the DNA repair defect, BRCA1/2 deficient tumor cells are more sensitive to PARP inhibitors (PARPi) through the mechanism of synthetic lethality. At present, several PAPRi targeting poly (ADP-ribose) polymerase (PARP) have been approved for ovarian cancer and breast cancer indications. However, PARPi resistance is ubiquitous in clinic. More than 40% BRCA1/2-deficient patients fail to respond to PARPi. In addition, lots of patients acquire PARPi resistance with prolonged oral administration of PARPi. Homologous recombination repair deficient (HRD), as an essential prerequisite of synthetic lethality, plays a vital role in killing tumor cells. Therefore, Homologous recombination repair restoration (HRR) becomes the predominant reason of PARPi resistance. Recently, it was reported that DNA replication fork protection also contributed to PARPi resistance in BRCA1/2-deficient cells and patients. Moreover, various factors, such as reversion mutations, epigenetic modification, restoration of ADP-ribosylation (PARylation) and pharmacological alteration lead to PARPi resistance as well. In this review, we reviewed the underlying mechanisms of PARP inhibitor resistance in detail and summarized the potential strategies to overcome PARPi resistance and increase PARPi sensitivity.
Keywords: Homologous recombination; PARPi; Resistance; Synthetic lethality.
Conflict of interest statement
The authors declare that there is no potential competing interest.
Figures


References
-
- Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16:35–42. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
