

Ethanol-mediated upregulation of APOA1 gene expression in HepG2 cells is independent of de novo lipid biosynthesis
- PMID: 32563265
- PMCID: PMC7306146
- DOI: 10.1186/s12944-020-01309-4
Ethanol-mediated upregulation of APOA1 gene expression in HepG2 cells is independent of de novo lipid biosynthesis
Abstract
Background: Moderate alcohol intake in human increases HDL-cholesterol, and has protective effects against cardiovascular disease (CVD). Although de novo lipid synthesis inhibitors are highly effective in lowering total and LDL-cholesterol they have only modest effects on raising HDL-C. A better understanding of the mechanism of ethanol-mediated HDL-C regulation could suggest new therapeutic approaches for CVD.
Methods: Human hepatoblastoma (HepG2) and colorectal epithelial adenocarcinoma (Caco-2) cells were incubated in the presence of varying concentrations of ethanol in the culture medium, with or without addition of de novo lipid synthesis (DNLS) inhibitors (atorvastatin and/or TOFA). ApoA1 protein was measured by Western blot, and RNA of lipid pathway genes APOA1, APOC3, APOA4, APOB100, HMGCR, LDLR, and SREBF2 by quantitative RT-PCR. Lipoproteins (VLDL, LDL, and HDL) and lipids were also monitored.
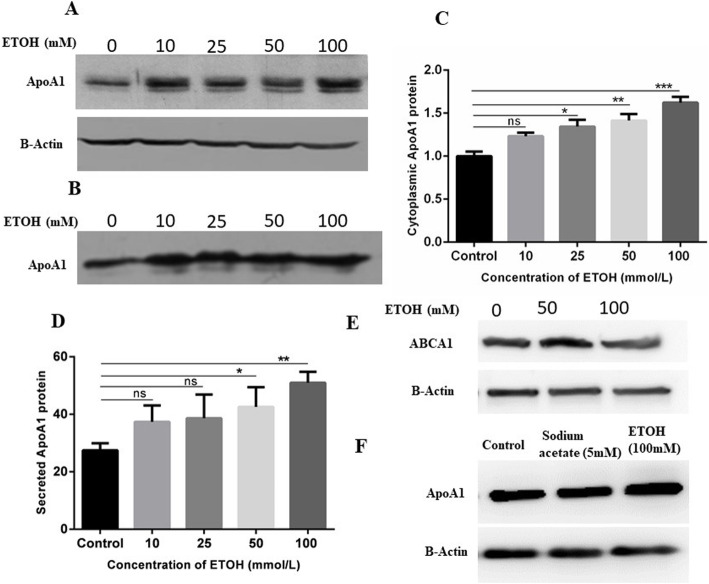
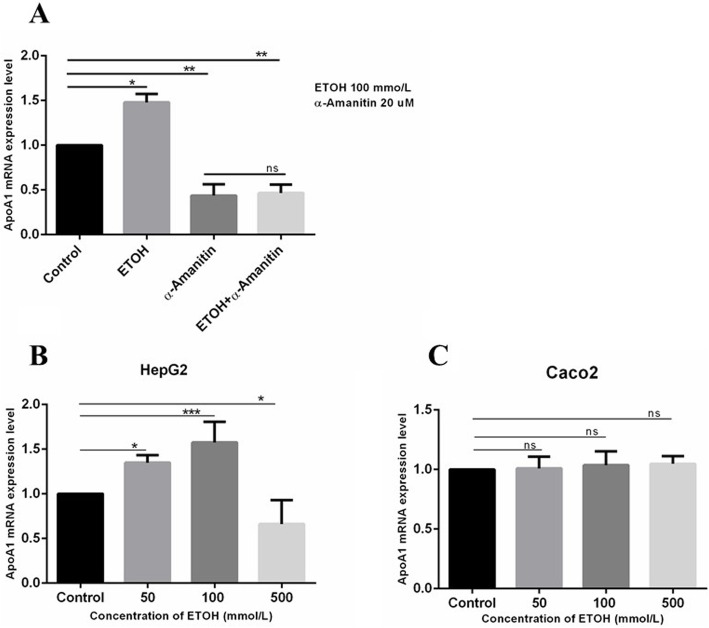
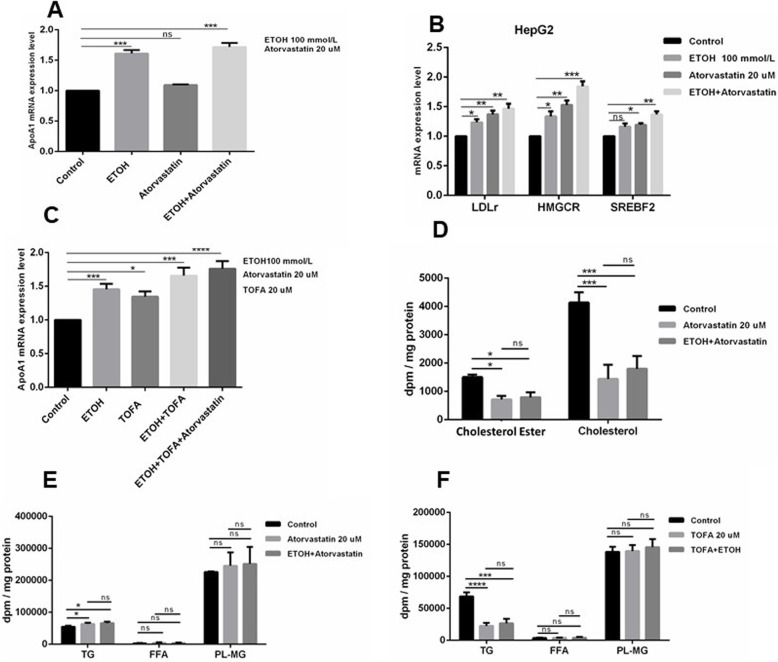
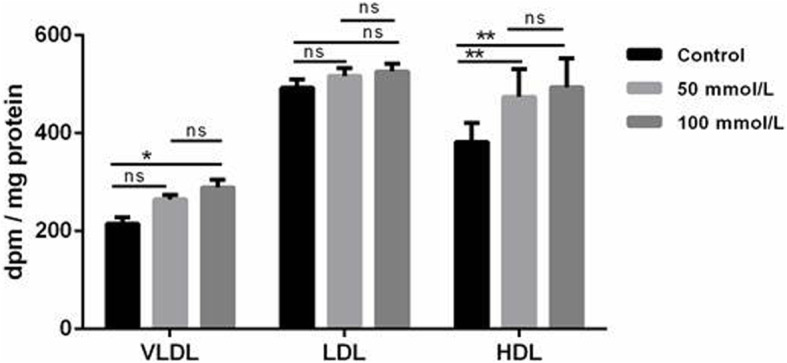
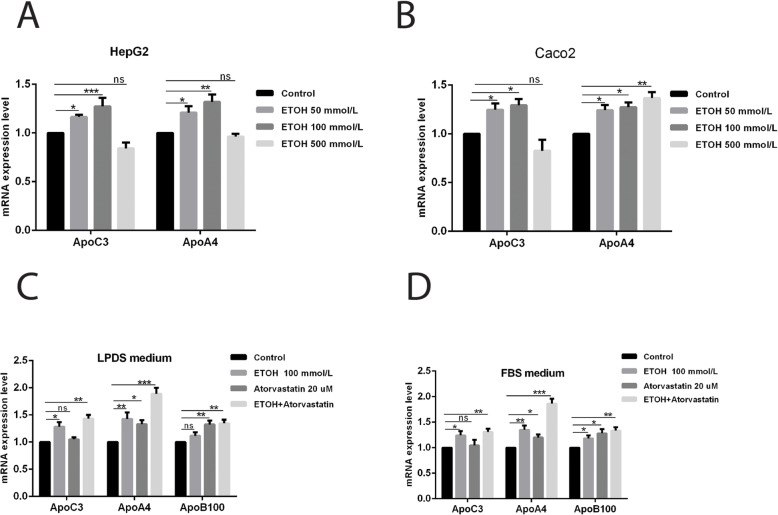
Results: Ethanol stimulated ApoA1 protein (both cytoplasmic and secreted) and APOA1 RNA levels in HepG2 cells in a dose sensitive way, with ~ 50% upregulation at 100 mM ethanol in the medium. The effect was not observed in intestinal-derived Caco-2 cells. DNLS inhibitors did not block the upregulation of ApoA1 RNA by ethanol; TOFA alone produced a modest increase in ApoA1 RNA. Ethanol had no effect on ABCA1 protein levels. Addition of ethanol to the cell medium led to modest increases in de novo synthesis of total cholesterol, cholesteryl esters and triglycerides, and as expected these increases were blocked when the lipid synthesis inhibitors were added. Ethanol stimulated a small increase in HDL and VLDL but not LDL synthesis. Ethanol in the cell medium also induced modest but measurable increases in the RNA of APOC3, APOA4, APOB, LDLR, and HMGCR genes. Unlike APOA1, induction of RNA from APOC3 and APOA4 was also observed in Caco-2 cells as well as HepG2 cells.
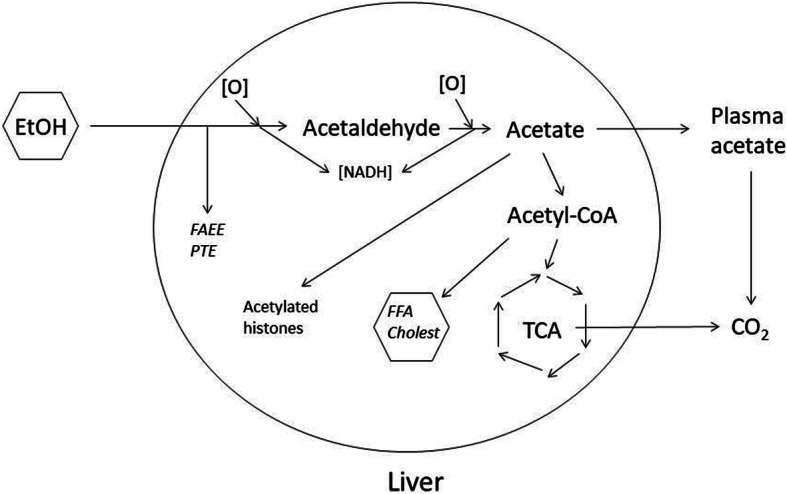
Conclusion: This study has verified the previously reported upregulation of APOA1 by exposure of HepG2, but not Caco-2 cells, to ethanol in the culture medium. It is shown for the first time that the effect is dependent on RNA polymerase II-mediated transcription, but not on de novo biosynthesis of cholesterol or fatty acids, and therefore is not a generalized metabolic response to ethanol exposure. Some other lipid pathway genes are also modulated by ethanol exposure of cells. The results reported here suggest that the proximal signaling molecule leading to increased APOA1 gene expression in response to ethanol exposure may be free acetate or acetyl-CoA.
Take home: Upregulation of ApoA1 gene expression in hepatoma cells in culture, upon exposure to moderate ethanol concentrations in the medium, occurs at the level of RNA and is not dependent on new cholesterol or fatty acid synthesis. The primary signaling molecule may be free acetate or acetyl-CoA. These results are important for understanding the mechanism by which moderate alcohol consumption leads to upregulation of serum HDL-cholesterol in humans, and suggests new approaches to targeting HDL as a risk factor for cardiovascular disease.
Keywords: APOA1; Alcohol; Apolipoprotein A1; Cardiovascular disease; Ethanol; HDL; HEPG2; Liver.
Conflict of interest statement
The authors state that they have no competing or conflict of interest.
Figures
Similar articles
-
Pitavastatin: novel effects on lipid parameters.Atheroscler Suppl. 2011 Nov;12(3):277-84. doi: 10.1016/S1567-5688(11)70887-X. Atheroscler Suppl. 2011. PMID: 22152282 Review.
-
Transcriptional suppression of human apolipoproteinA4 and apolipoproteinC3 genes by phorbol myristate acetate in hepatic and intestinal cells.Biomed Mater Eng. 2014;24(1):877-84. doi: 10.3233/BME-130880. Biomed Mater Eng. 2014. PMID: 24211975
-
Interactions between the apolipoprotein a1/c3/a5 haplotypes and alcohol consumption on serum lipid levels.Alcohol Clin Exp Res. 2013 Feb;37(2):234-43. doi: 10.1111/j.1530-0277.2012.01918.x. Epub 2012 Aug 24. Alcohol Clin Exp Res. 2013. PMID: 22924697
-
An experimental study on amelioration of dyslipidemia-induced atherosclesis by Clematichinenoside through regulating Peroxisome proliferator-activated receptor-α mediated apolipoprotein A-I, A-II and C-III.Eur J Pharmacol. 2015 Aug 15;761:362-74. doi: 10.1016/j.ejphar.2015.04.015. Epub 2015 May 12. Eur J Pharmacol. 2015. PMID: 25979856
-
The responses of different dosages of egg consumption on blood lipid profile: An updated systematic review and meta-analysis of randomized clinical trials.J Food Biochem. 2020 Aug;44(8):e13263. doi: 10.1111/jfbc.13263. Epub 2020 Jun 11. J Food Biochem. 2020. PMID: 32524644
Cited by
-
U-shaped relationship between apolipoprotein A1 levels and mortality risk in men and women.Eur J Prev Cardiol. 2023 Mar 1;30(4):293-304. doi: 10.1093/eurjpc/zwac263. Eur J Prev Cardiol. 2023. PMID: 36351048 Free PMC article.
-
Activation/Inhibition of Gene Expression Caused by Alcohols: Relationship with the Viscoelastic Property of a DNA Molecule.Polymers (Basel). 2022 Dec 28;15(1):149. doi: 10.3390/polym15010149. Polymers (Basel). 2022. PMID: 36616499 Free PMC article.
-
The Lipidomic Profile Discriminates Between MASLD and MetALD.Aliment Pharmacol Ther. 2025 Apr;61(8):1357-1371. doi: 10.1111/apt.70012. Epub 2025 Feb 11. Aliment Pharmacol Ther. 2025. PMID: 39935287 Free PMC article.
-
Amino Acid and Fatty Acid Metabolism Disorders Trigger Oxidative Stress and Inflammatory Response in Excessive Dietary Valine-Induced NAFLD of Laying Hens.Front Nutr. 2022 Apr 12;9:849767. doi: 10.3389/fnut.2022.849767. eCollection 2022. Front Nutr. 2022. PMID: 35495903 Free PMC article.
-
System Biology Investigation Revealed Lipopolysaccharide and Alcohol-Induced Hepatocellular Carcinoma Resembled Hepatitis B Virus Immunobiology and Pathogenesis.Int J Mol Sci. 2023 Jul 6;24(13):11146. doi: 10.3390/ijms241311146. Int J Mol Sci. 2023. PMID: 37446321 Free PMC article.
References
-
- Joseph P, Leong D, McKee M, Anand SS, Schwalm JD, Teo K, et al. Reducing the global burden of cardiovascular disease, part 1: the epidemiology and Risk Factors. Circ Res. 2017;121(6):677–94. - PubMed
-
- Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. Jama. 1986;256(20):2835–2838. - PubMed
-
- Sharrett AR, Ballantyne CM, Coady SA, Heiss G, Sorlie PD, Catellier D, et al. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: the atherosclerosis Risk in communities (ARIC) study. Circulation. 2001;104(10):1108–1113. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
