

Metabolic liver disease in diabetes - From mechanisms to clinical trials
- PMID: 32569680
- PMCID: PMC7305712
- DOI: 10.1016/j.metabol.2020.154299
Metabolic liver disease in diabetes - From mechanisms to clinical trials
Abstract
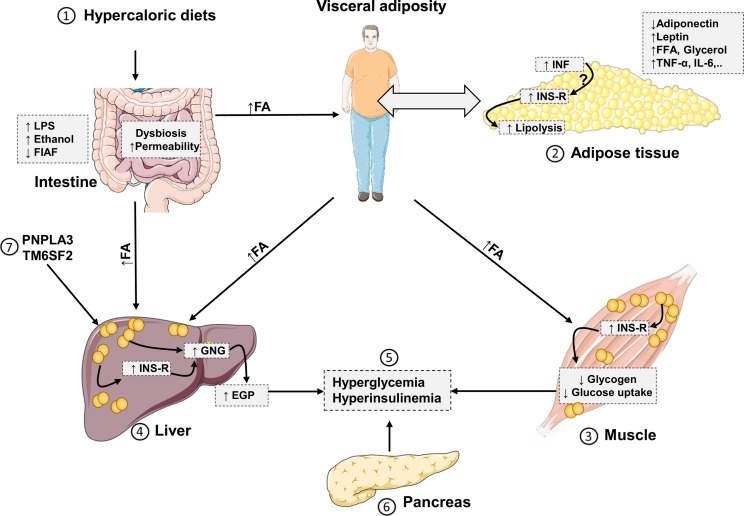
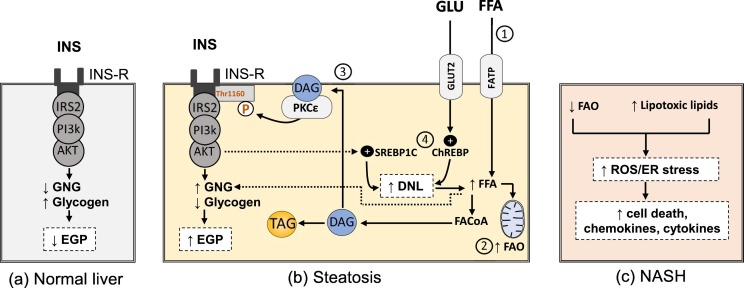
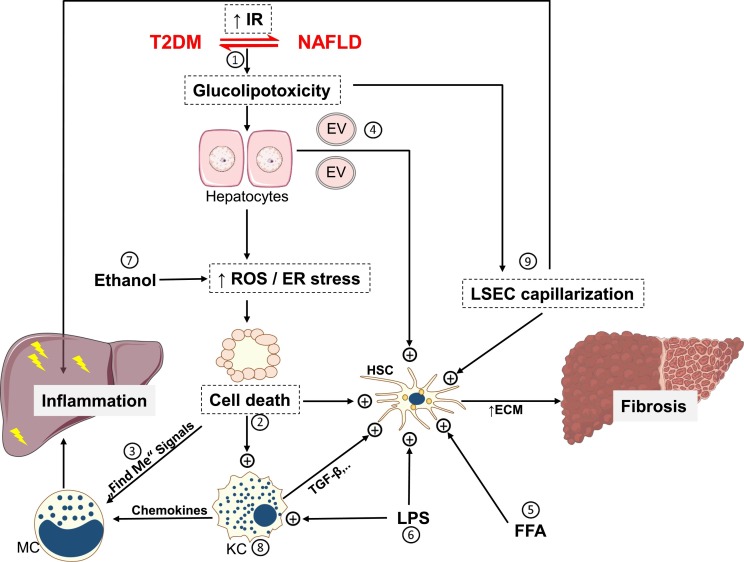
Non-alcoholic fatty liver disease (NAFLD) comprises fatty liver (steatosis), non-alcoholic steatohepatitis (NASH) and fibrosis/cirrhosis and may lead to end-stage liver failure or hepatocellular carcinoma. NAFLD is tightly associated with the most frequent metabolic disorders, such as obesity, metabolic syndrome, and type 2 diabetes mellitus (T2DM). Both multisystem diseases share several common mechanisms. Alterations of tissue communications include excessive lipid and later cytokine release by dysfunctional adipose tissue, intestinal dysbiosis and ectopic fat deposition in skeletal muscle. On the hepatocellular level, this leads to insulin resistance due to abnormal lipid handling and mitochondrial function. Over time, cellular oxidative stress and activation of inflammatory pathways, again supported by multiorgan crosstalk, determine NAFLD progression. Recent studies show that particularly the severe insulin resistant diabetes (SIRD) subgroup (cluster) associates with NAFLD and its accelerated progression and increases the risk of diabetes-related cardiovascular and kidney diseases, underpinning the critical role of insulin resistance. Consequently, lifestyle modification and certain drug classes used to treat T2DM have demonstrated effectiveness for treating NAFLD, but also some novel therapeutic concepts may be beneficial for both NAFLD and T2DM. This review addresses the bidirectional relationship between mechanisms underlying T2DM and NAFLD, the relevance of novel biomarkers for improving the diagnostic modalities and the identification of subgroups at specific risk of disease progression. Also, the role of metabolism-related drugs in NAFLD is discussed in light of the recent clinical trials. Finally, this review highlights some challenges to be addressed by future studies on NAFLD in the context of T2DM.
Keywords: Biomarkers; Clinical trials; Fatty liver; Glucose-lowering drugs; Insulin resistance; Type 2 diabetes.
Copyright © 2020 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of competing interest BD, SK and KP declare no conflicts of interest. MR is on the scientific advisory boards of Allergan, Astra-Zeneca, Bristol-Myers Squibb, Eli Lilly, Gilead Sciences, Inventiva, Intercept Pharma, Novartis, NovoNordisk, Servier Laboratories, Target Pharmasolutions, and Terra Firma and receives investigator-initiated support from Boehringer Ingelheim, Nutricia/Danone and Sanofi–Aventis.
Figures
References
-
- European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO clinical practice guidelines for the management of non-alcoholic fatty liver disease. J Hepatol 2016;64:1388–402. 10.1016/j.jhep.2015.11.004. - DOI - PubMed
-
- Lammert F. Clinical genomics of NAFLD. The liver, John Wiley & Sons, Ltd; 2020, p. 509–20. 10.1002/9781119436812.ch41. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
