

Gene expression predictions and networks in natural populations supports the omnigenic theory
- PMID: 32571208
- PMCID: PMC7310122
- DOI: 10.1186/s12864-020-06809-2
Gene expression predictions and networks in natural populations supports the omnigenic theory
Abstract
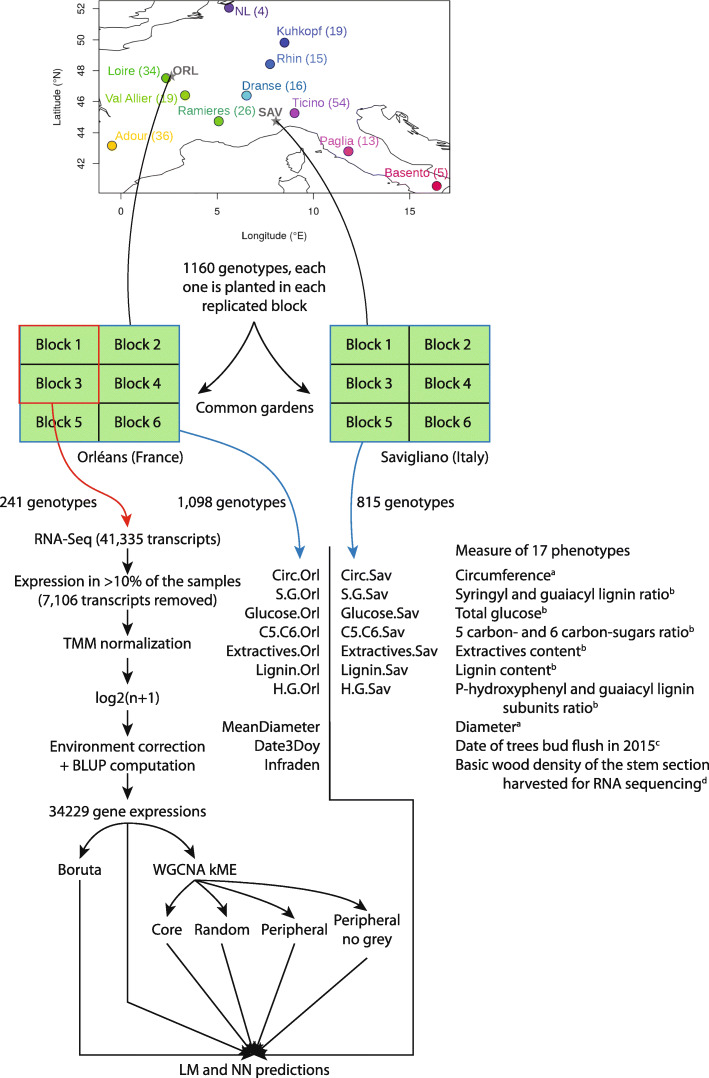
Background: Recent literature on the differential role of genes within networks distinguishes core from peripheral genes. If previous works have shown contrasting features between them, whether such categorization matters for phenotype prediction remains to be studied.
Results: We measured 17 phenotypic traits for 241 cloned genotypes from a Populus nigra collection, covering growth, phenology, chemical and physical properties. We also sequenced RNA for each genotype and built co-expression networks to define core and peripheral genes. We found that cores were more differentiated between populations than peripherals while being less variable, suggesting that they have been constrained through potentially divergent selection. We also showed that while cores were overrepresented in a subset of genes statistically selected for their capacity to predict the phenotypes (by Boruta algorithm), they did not systematically predict better than peripherals or even random genes.
Conclusion: Our work is the first attempt to assess the importance of co-expression network connectivity in phenotype prediction. While highly connected core genes appear to be important, they do not bear enough information to systematically predict better quantitative traits than other gene sets.
Keywords: Boruta; Core; Machine learning; Peripheral; Populus nigra.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- Mackay TFC, Stone Ea, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10(8):565–77. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
