

A community-maintained standard library of population genetic models
- PMID: 32573438
- PMCID: PMC7438115
- DOI: 10.7554/eLife.54967
A community-maintained standard library of population genetic models
Abstract
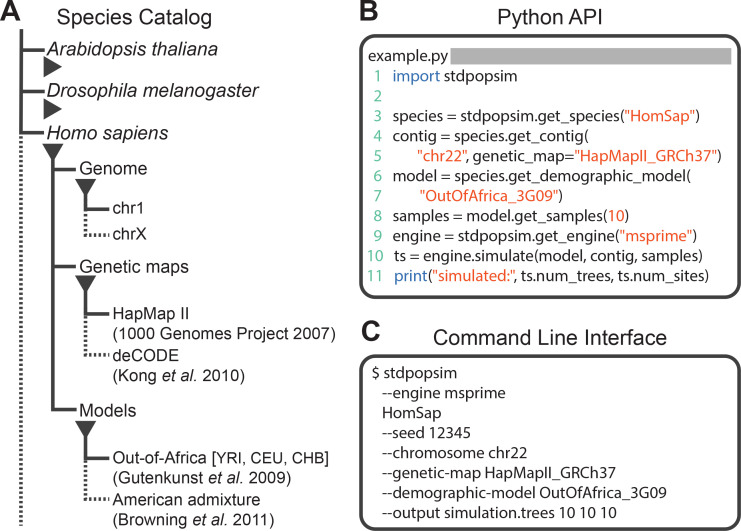
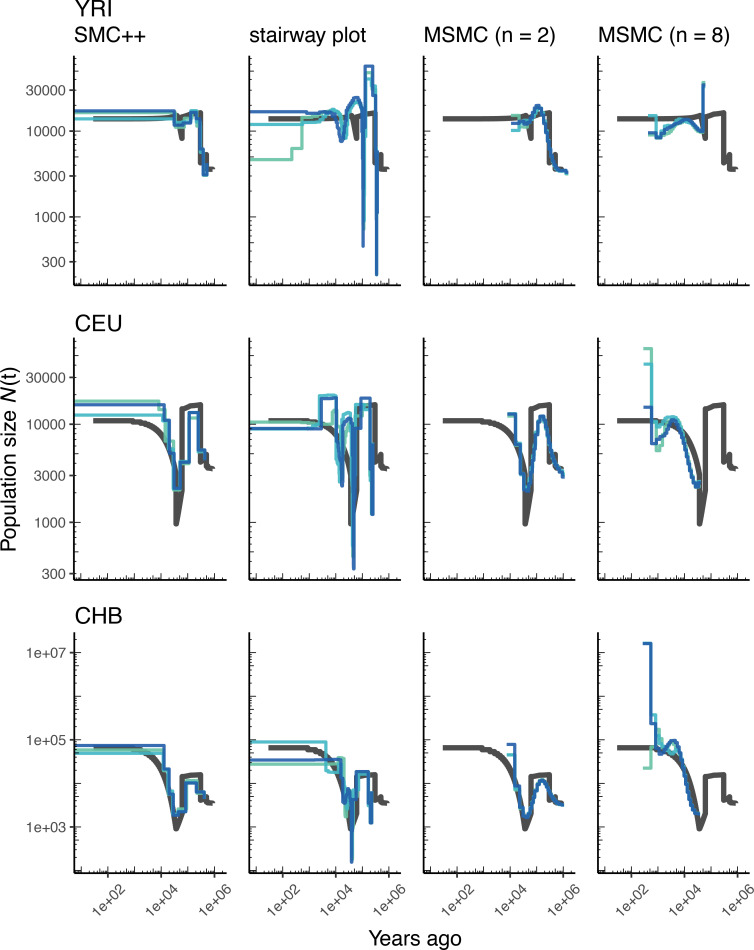
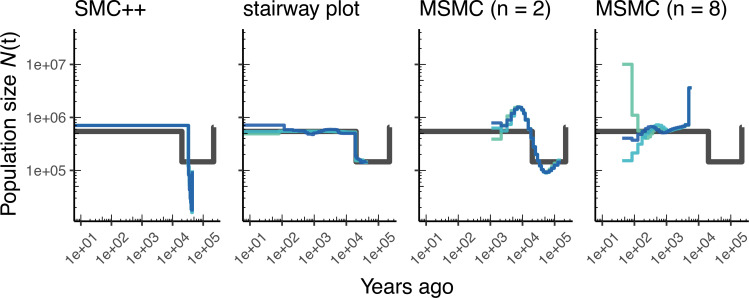
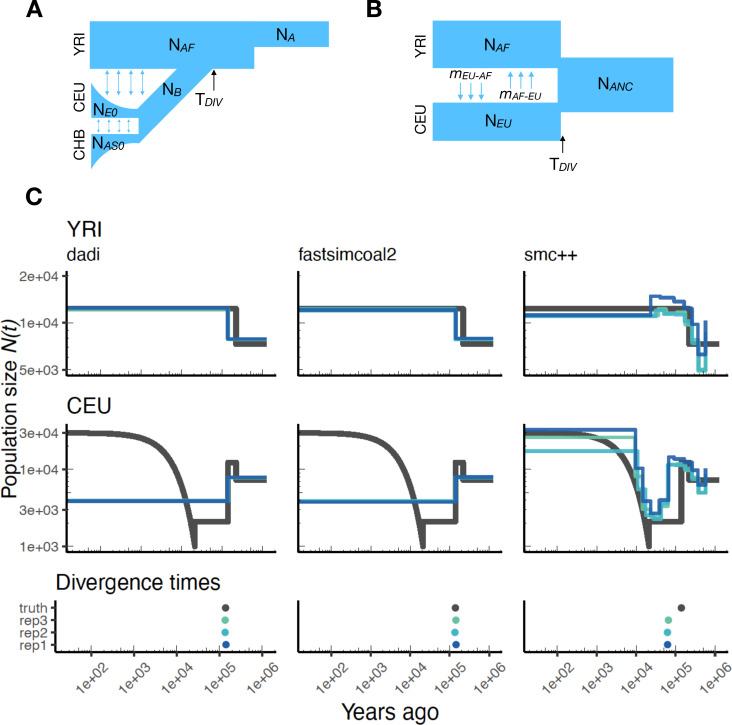
The explosion in population genomic data demands ever more complex modes of analysis, and increasingly, these analyses depend on sophisticated simulations. Recent advances in population genetic simulation have made it possible to simulate large and complex models, but specifying such models for a particular simulation engine remains a difficult and error-prone task. Computational genetics researchers currently re-implement simulation models independently, leading to inconsistency and duplication of effort. This situation presents a major barrier to empirical researchers seeking to use simulations for power analyses of upcoming studies or sanity checks on existing genomic data. Population genetics, as a field, also lacks standard benchmarks by which new tools for inference might be measured. Here, we describe a new resource, stdpopsim, that attempts to rectify this situation. Stdpopsim is a community-driven open source project, which provides easy access to a growing catalog of published simulation models from a range of organisms and supports multiple simulation engine backends. This resource is available as a well-documented python library with a simple command-line interface. We share some examples demonstrating how stdpopsim can be used to systematically compare demographic inference methods, and we encourage a broader community of developers to contribute to this growing resource.
Keywords: computational biology; evolutionary biology; human; open source; reproducibility; simulation; systems biology.
© 2020, Adrion et al.
Conflict of interest statement
JA, CC, ND, JG, AG, GG, CK, AR, GT, FB, JC, RC, AD, IG, BK, PM, EN, DO, FR, TS, SG, RG, KL, PR, DS, AS, JK, AK No competing interests declared, PM Reviewing editor, eLife
Figures
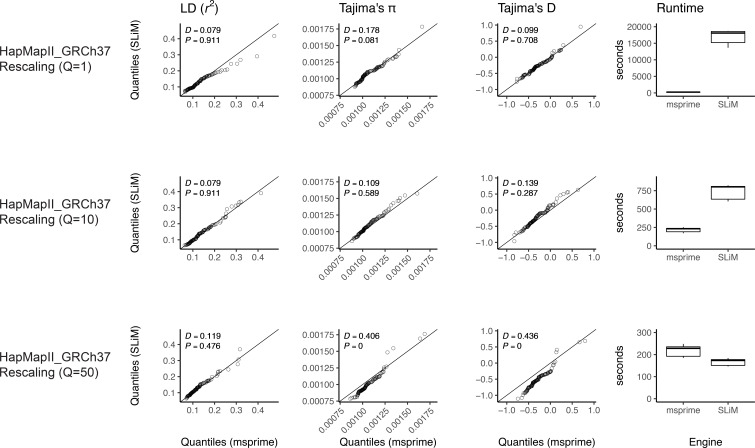
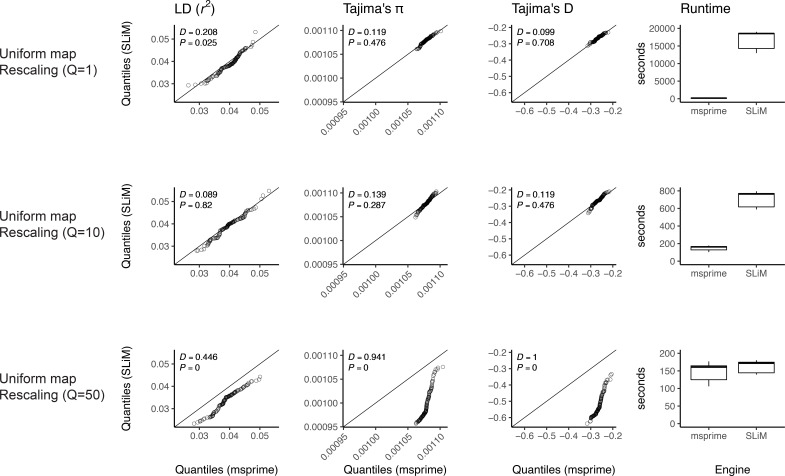
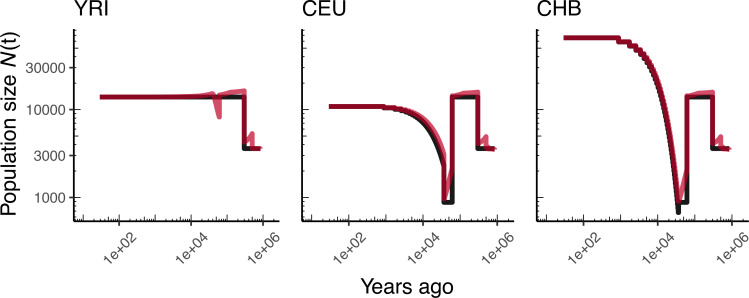
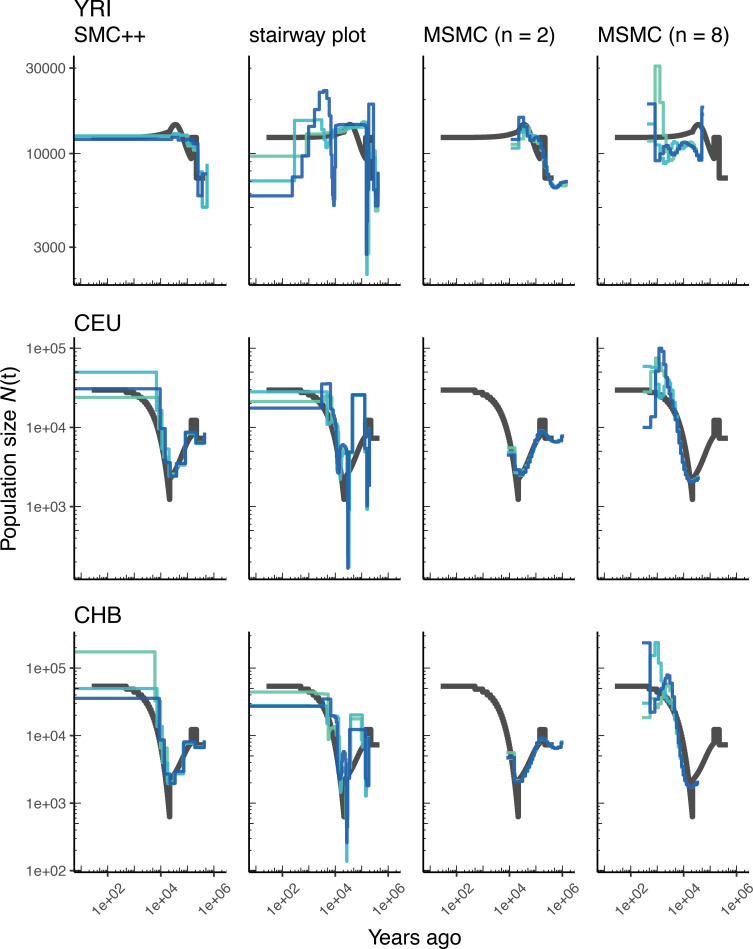
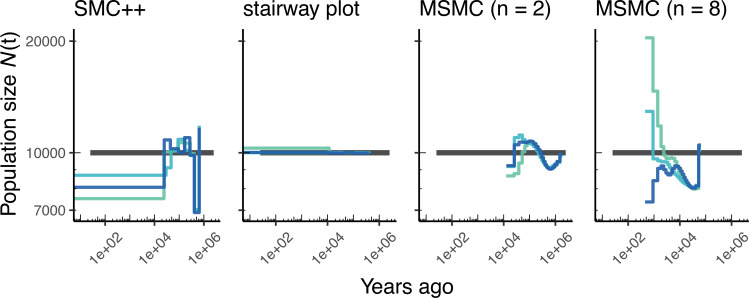
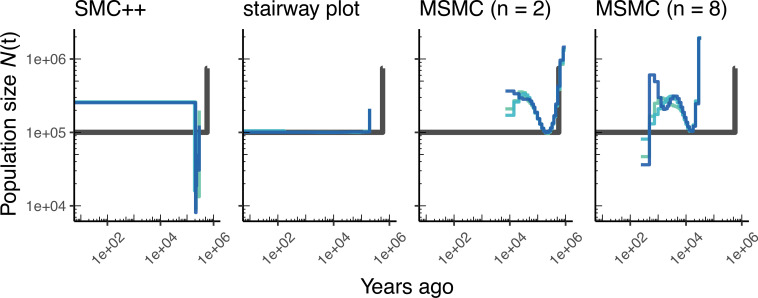
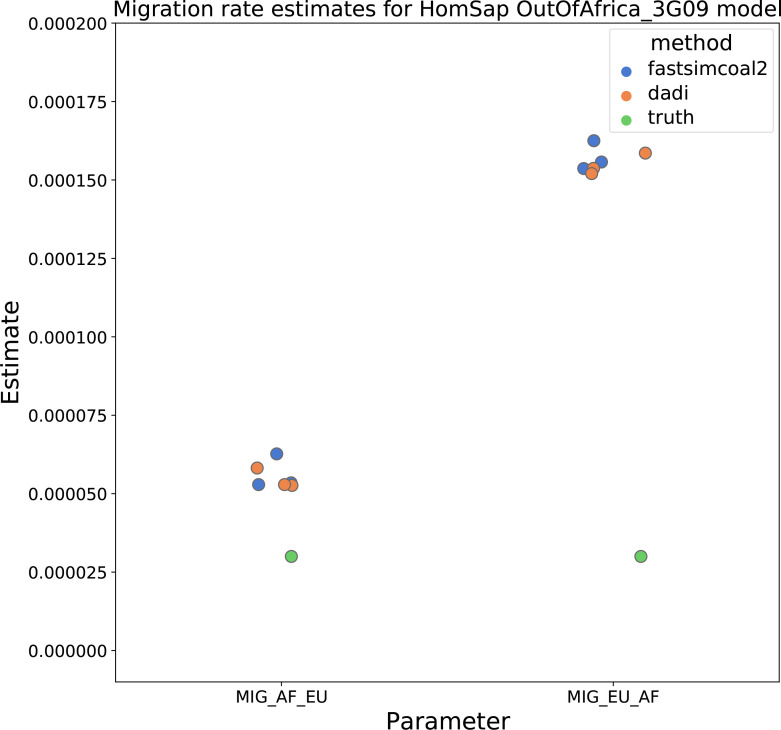
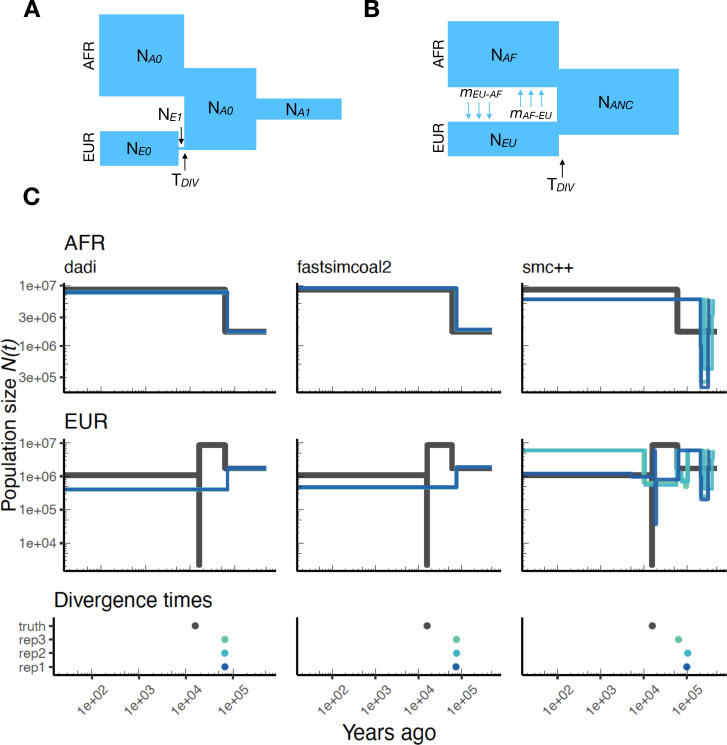
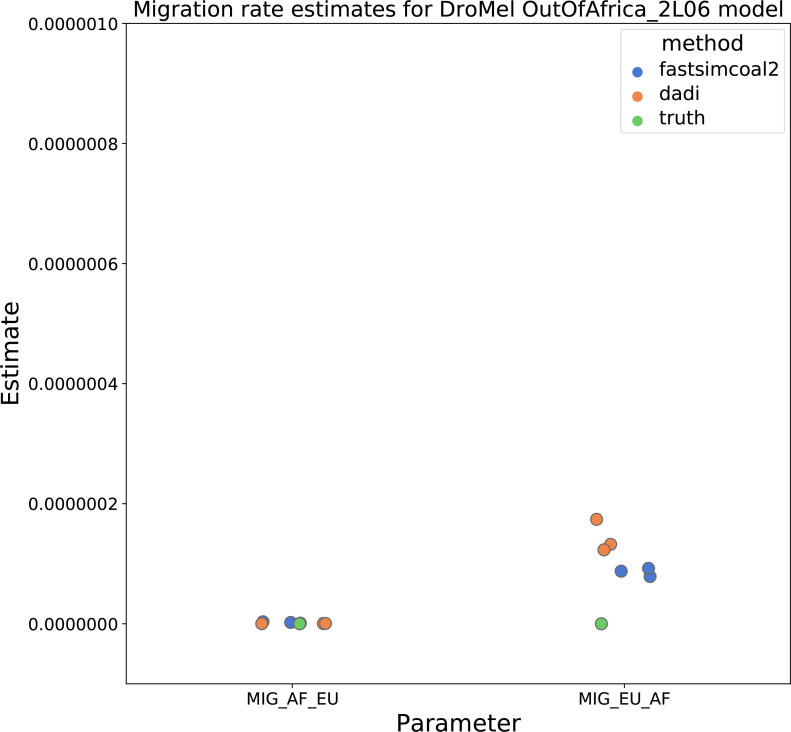
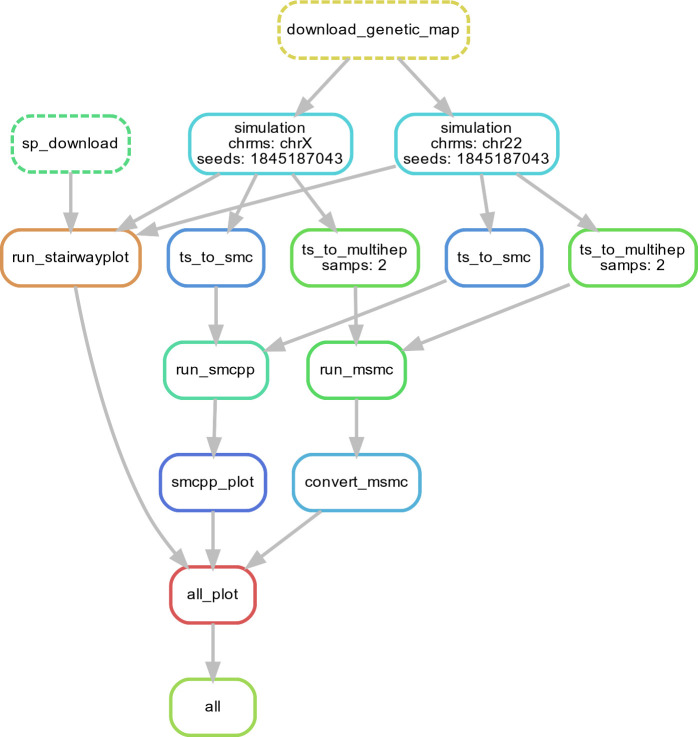
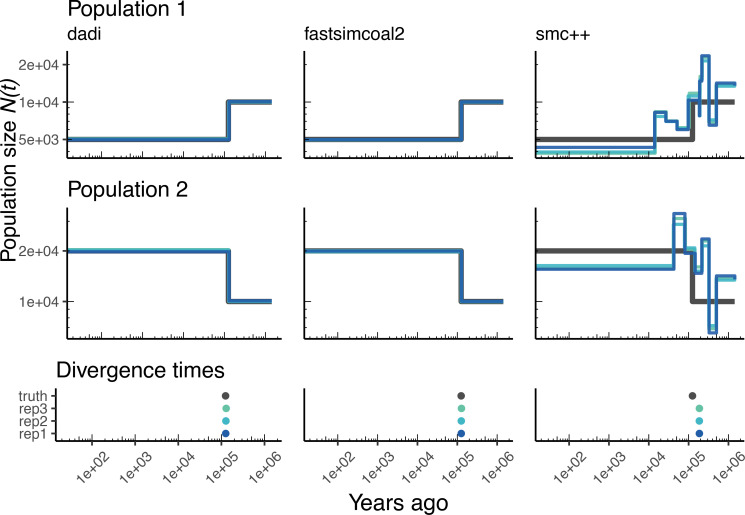
Comment in
-
Standardizing population genetics simulations.Nat Methods. 2020 Sep;17(9):876. doi: 10.1038/s41592-020-0951-4. Nat Methods. 2020. PMID: 32873980 No abstract available.
References
-
- Beichman AC, Huerta-Sanchez E, Lohmueller KE. Using genomic data to infer historic population dynamics of nonmodel organisms. Annual Review of Ecology, Evolution, and Systematics. 2018;49:433–456. doi: 10.1146/annurev-ecolsys-110617-062431. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
