

Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia
- PMID: 32573726
- PMCID: PMC7717479
- DOI: 10.1182/blood.2019004560
Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia
Abstract
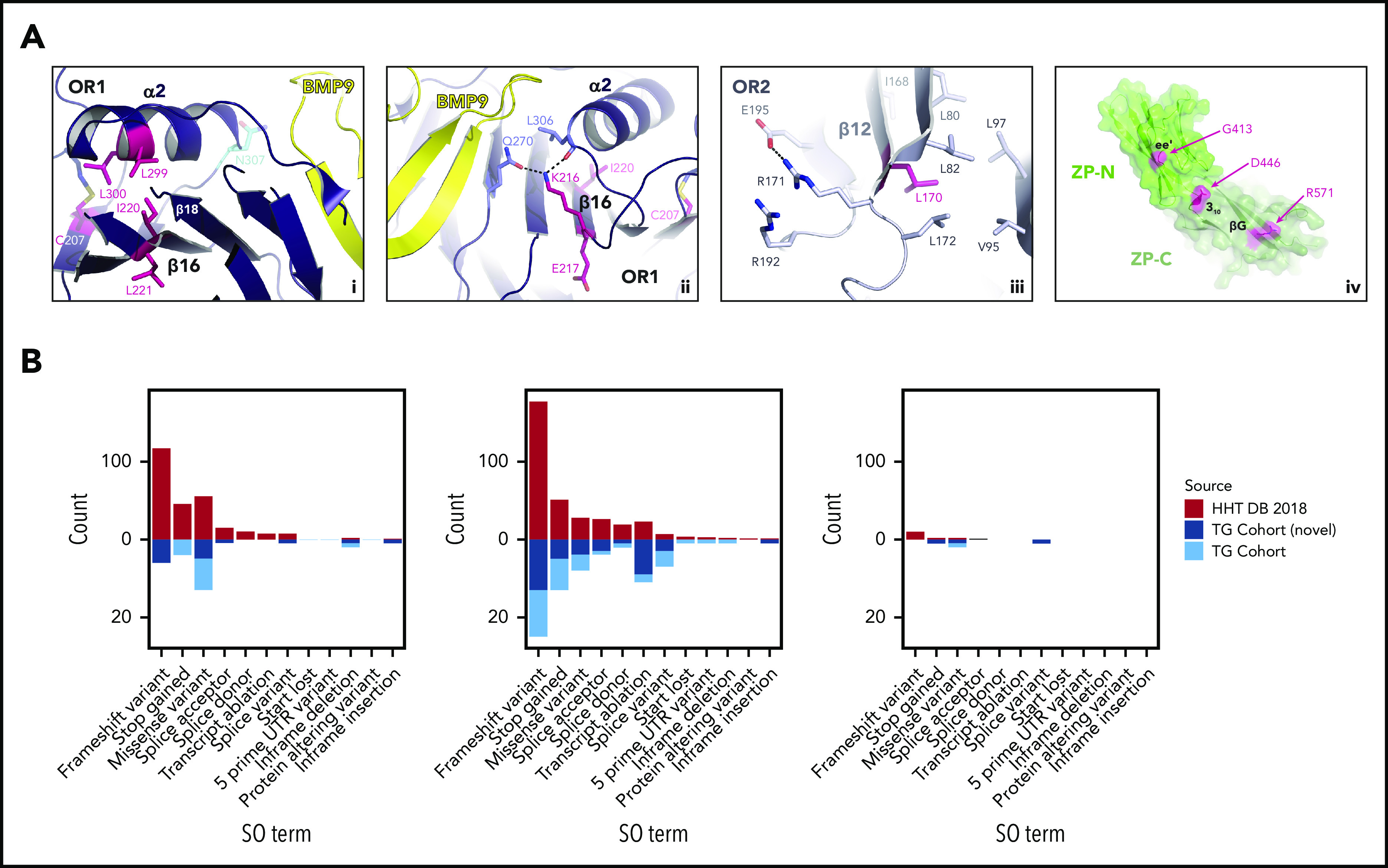
Hereditary hemorrhagic telangiectasia (HHT) is an autosomal dominant vascular dysplasia. Care delivery for HHT patients is impeded by the need for laborious, repeated phenotyping and gaps in knowledge regarding the relationships between causal DNA variants in ENG, ACVRL1, SMAD4 and GDF2, and clinical manifestations. To address this, we analyzed DNA samples from 183 previously uncharacterized, unrelated HHT and suspected HHT cases using the ThromboGenomics high-throughput sequencing platform. We identified 127 rare variants across 168 heterozygous genotypes. Applying modified American College of Medical Genetics and Genomics Guidelines, 106 variants were classified as pathogenic/likely pathogenic and 21 as nonpathogenic (variant of uncertain significance/benign). Unlike the protein products of ACVRL1 and SMAD4, the extracellular ENG amino acids are not strongly conserved. Our inferences of the functional consequences of causal variants in ENG were therefore informed by the crystal structure of endoglin. We then compared the accuracy of predictions of the causal gene blinded to the genetic data using 2 approaches: subjective clinical predictions and statistical predictions based on 8 Human Phenotype Ontology terms. Both approaches had some predictive power, but they were insufficiently accurate to be used clinically, without genetic testing. The distributions of red cell indices differed by causal gene but not sufficiently for clinical use in isolation from genetic data. We conclude that parallel sequencing of the 4 known HHT genes, multidisciplinary team review of variant calls in the context of detailed clinical information, and statistical and structural modeling improve the prognostication and treatment of HHT.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures





References
-
- VASCERN-HHT European Reference Network For Vascular Diseases Hereditary hemorrhagic telangiectasia. www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=236. Accessed 24 August 2020.
-
- Bideau A, Brunet G, Heyer E, Plauchu H, Robert J-M. An abnormal concentration of cases of Rendu-Osler disease in the Valserine valley of the French Jura: a genealogical and demographic study. Ann Hum Biol. 1992;19(3):233-247. - PubMed
-
- Dakeishi M, Shioya T, Wada Y, et al. . Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum Mutat. 2002;19(2):140-148. - PubMed
-
- Kjeldsen AD, Vase P, Green A. Hereditary haemorrhagic telangiectasia: a population-based study of prevalence and mortality in Danish patients. J Intern Med. 1999;245(1):31-39. - PubMed
-
- Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. The UK prevalence of hereditary haemorrhagic telangiectasia and its association with sex, socioeconomic status and region of residence: a population-based study. Thorax. 2014;69(2):161-167. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- Wellcome Trust/United Kingdom
- R35 HL140019/HL/NHLBI NIH HHS/United States
- TF/037257/Wellcome Trust/United Kingdom
- AF/053286/Wellcome Trust/United Kingdom
- PG/2000067/British Heart Foundation/United Kingdom
- FS/04/089/British Heart Foundation/United Kingdom
- PG/09/041/27515/British Heart Foundation/United Kingdom
- RBAG/342/Wellcome Trust/United Kingdom
- RBAG/245/British Heart Foundation/United Kingdom
- RBAG/208/British Heart Foundation/United Kingdom
- RBAG/226/British Heart Foundation/United Kingdom
- RBAG/285/Medical Research Council/United Kingdom
- RBAG/295/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
