

Integrating Immunotherapy and Targeted Therapy in Cancer Treatment: Mechanistic Insights and Clinical Implications
- PMID: 32576627
- PMCID: PMC7641965
- DOI: 10.1158/1078-0432.CCR-19-2300
Integrating Immunotherapy and Targeted Therapy in Cancer Treatment: Mechanistic Insights and Clinical Implications
Abstract
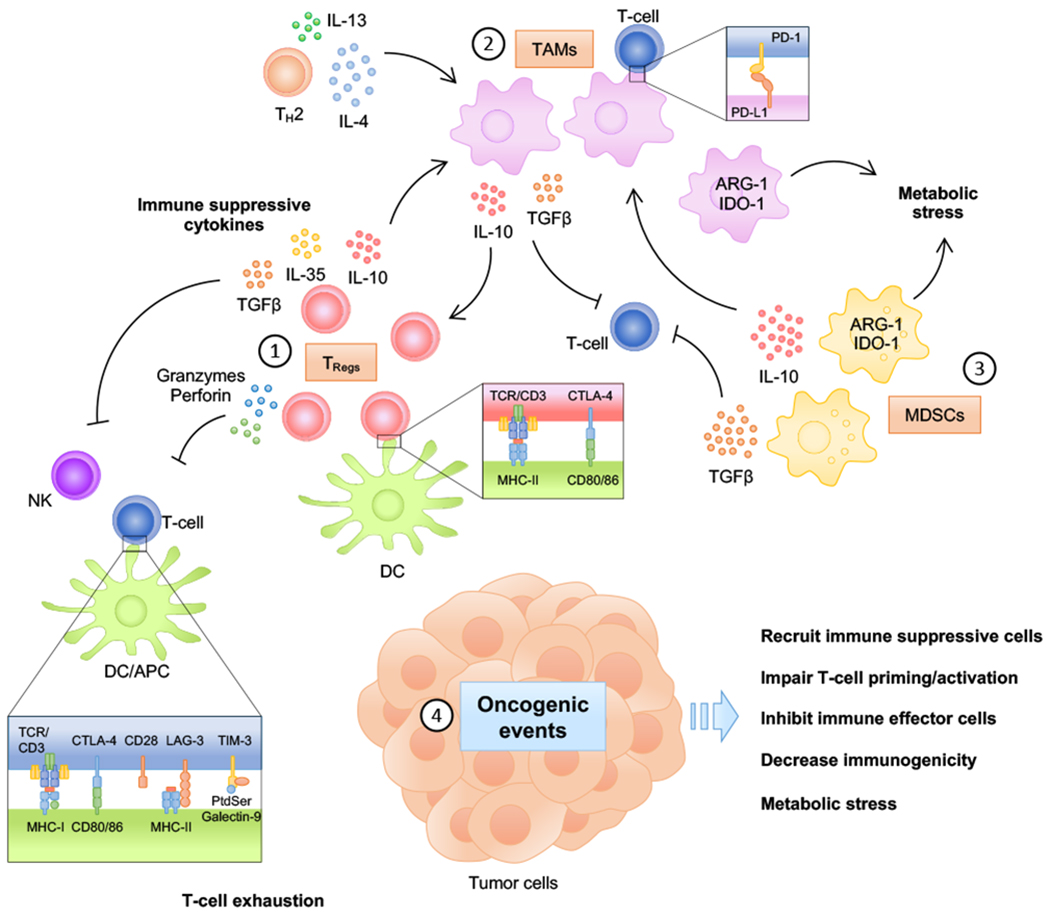
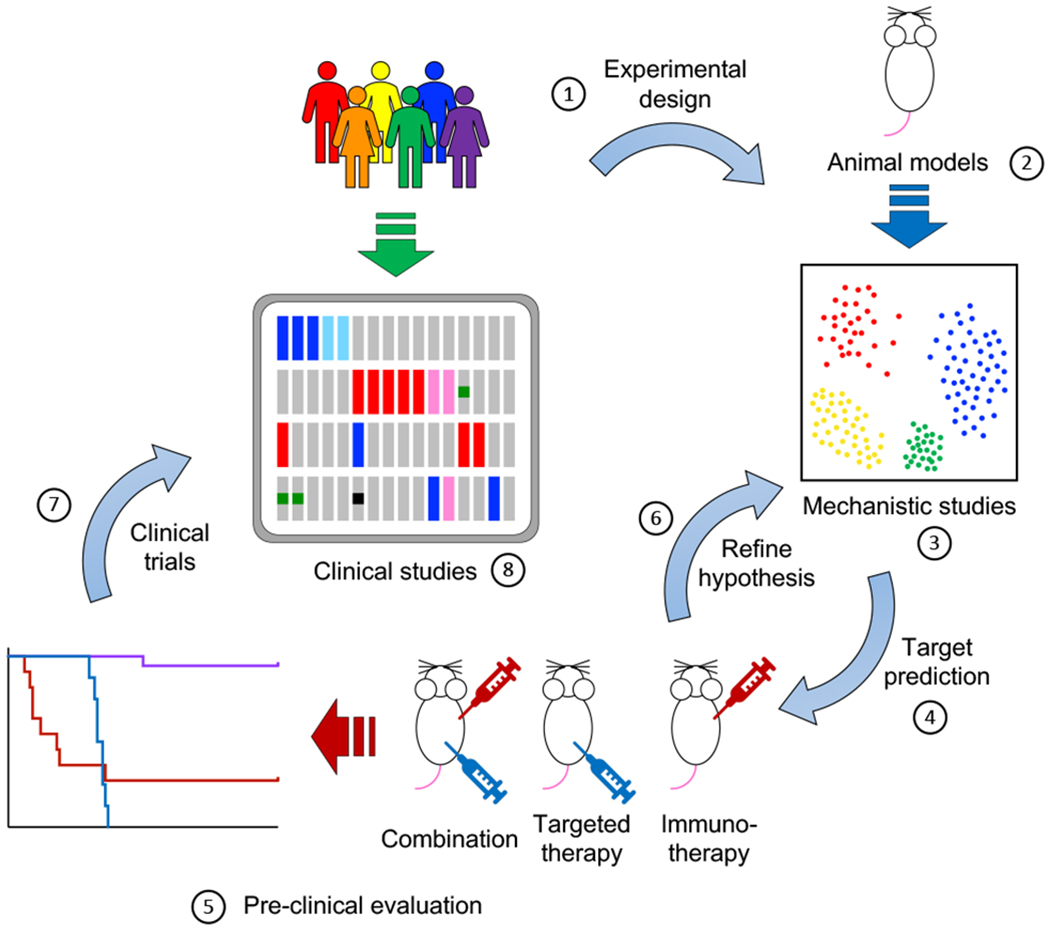
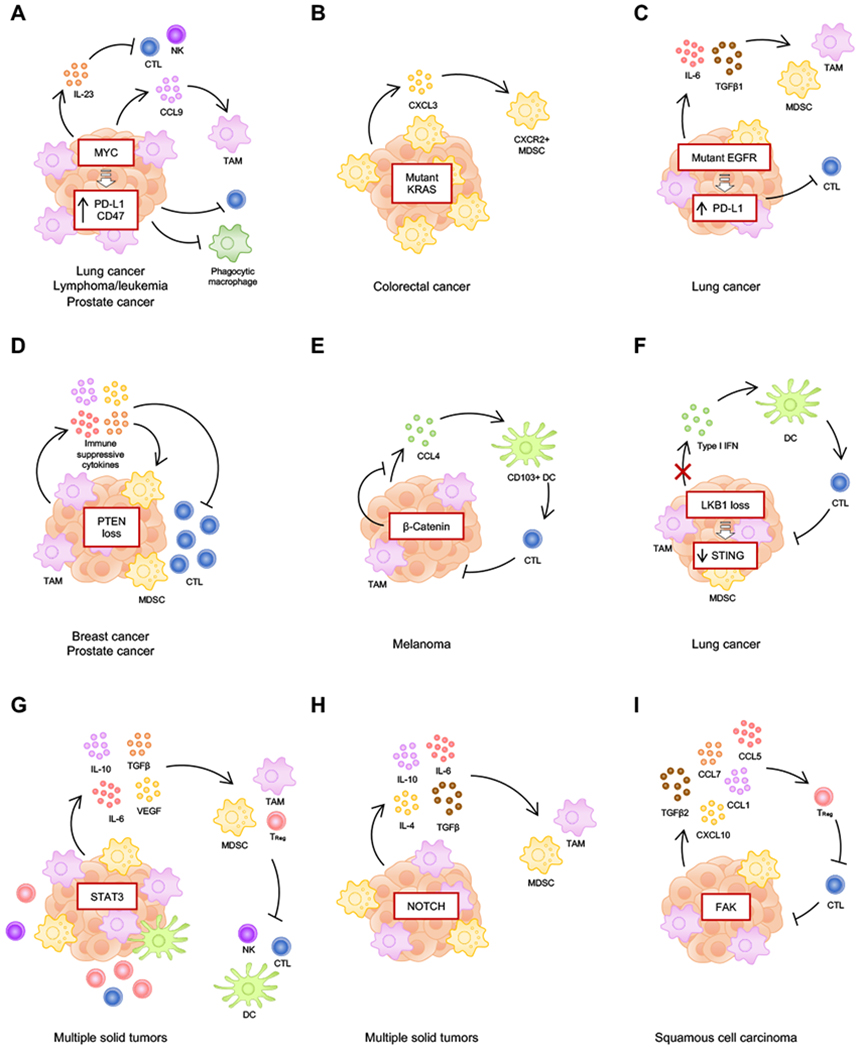
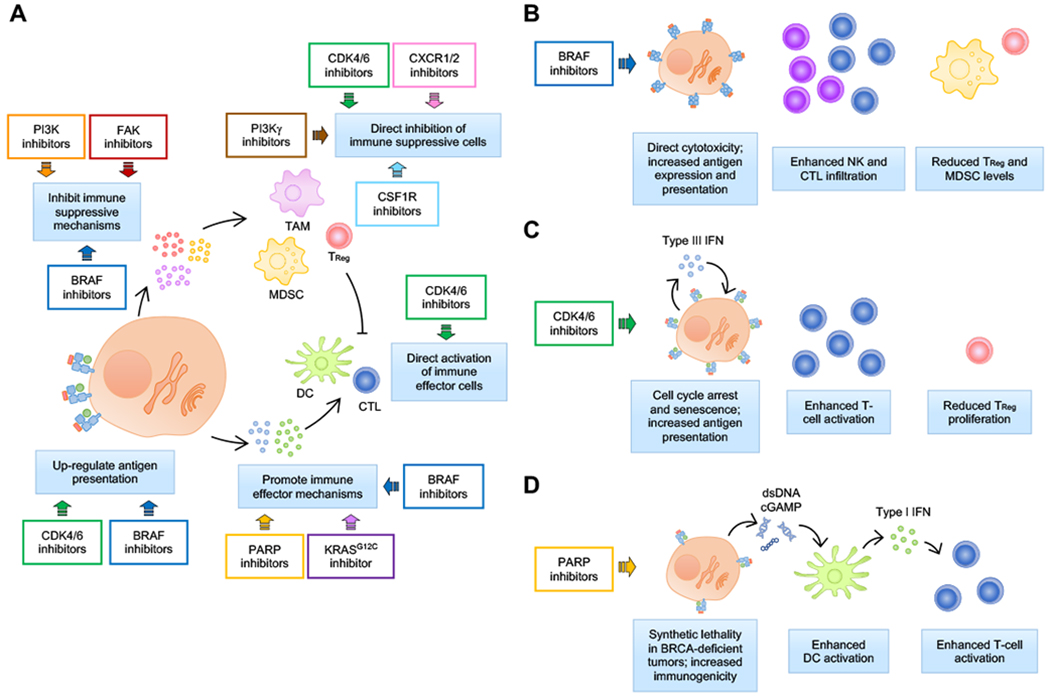
Small-molecule targeted therapies have demonstrated outstanding potential in the clinic. These drugs are designed to minimize adverse effects by selectively attacking cancer cells while exerting minimal damage to normal cells. Although initial response to targeted therapies may be high, yielding positive response rates and often improving survival for an important percentage of patients, resistance often limits long-term effectiveness. On the other hand, immunotherapy has demonstrated durable results, yet for a limited number of patients. Growing evidence indicates that some targeted agents can modulate different components of the antitumor immune response. These include immune sensitization by inhibiting tumor cell-intrinsic immune evasion programs or enhancing antigenicity, as well as direct effects on immune effector and immunosuppressive cells. The combination of these two approaches, therefore, has the potential to result in synergistic and durable outcomes for patients. In this review, we focus on the latest advances on integrating immunotherapy with small-molecule targeted inhibitors. In particular, we discuss how specific oncogenic events differentially affect immune response, and the implications of these findings on the rational design of effective combinations of immunotherapy and targeted therapies.
©2020 American Association for Cancer Research.
Conflict of interest statement
Disclosure of potential conflicts of interest
J.S.B. is a scientific consultant for Geode Therapeutics. J.S.B. and J.J.Z. are co-inventors of DFCI 2180.001 (DFS-166.25). Q.W. is a scientific consultant for Crimson Biopharm. Q.W. and J.J.Z. are co-inventors of DFCI 2409.001 (DFS-203.60). J.J.Z. is a founder and director of Crimson Biopharm and Geode Therapeutics.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
