

Joint reconstruction of cis-regulatory interaction networks across multiple tissues using single-cell chromatin accessibility data
- PMID: 32578841
- PMCID: PMC8138825
- DOI: 10.1093/bib/bbaa120
Joint reconstruction of cis-regulatory interaction networks across multiple tissues using single-cell chromatin accessibility data
Abstract
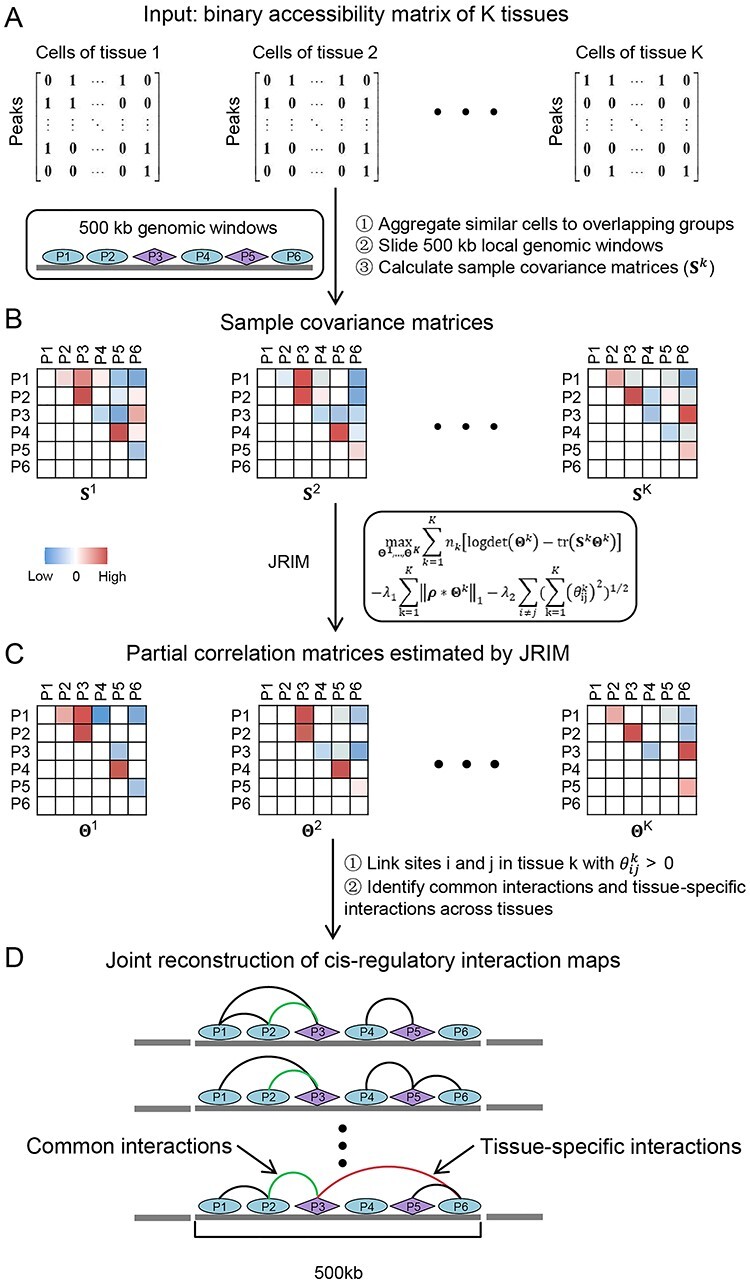
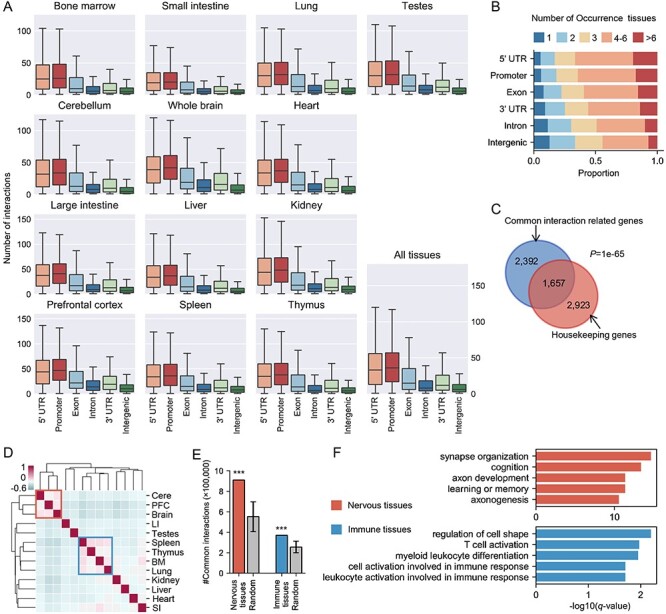
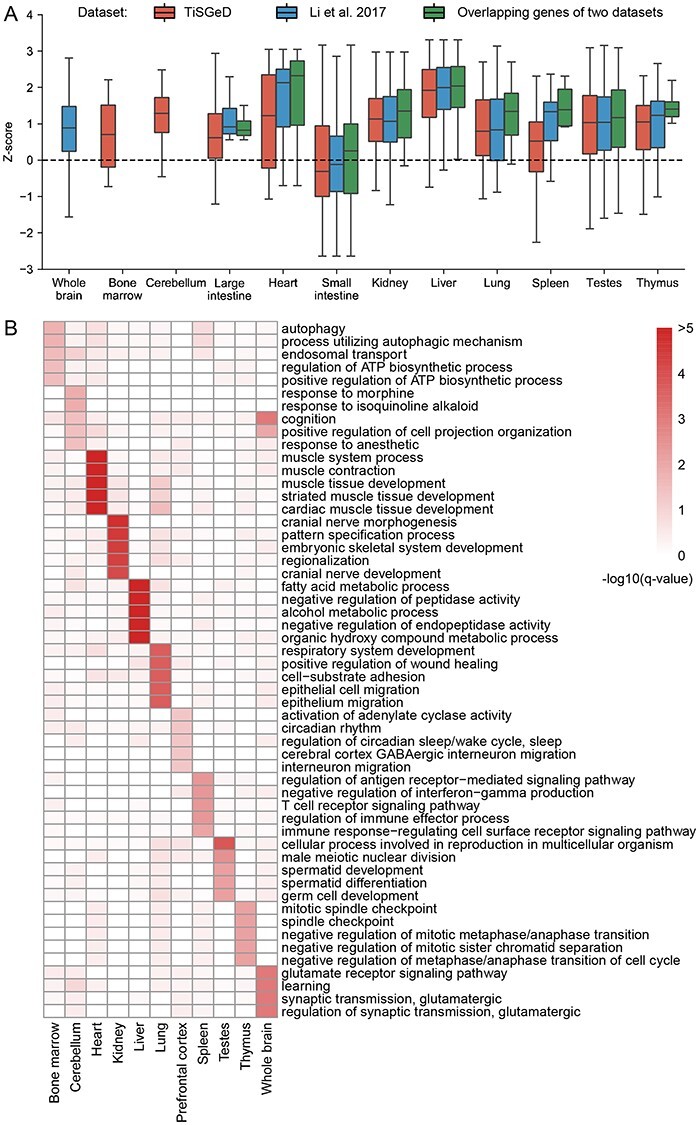
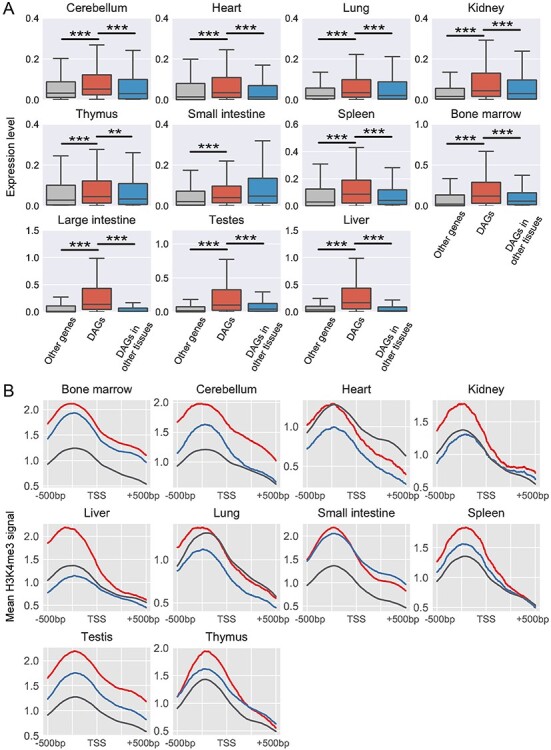
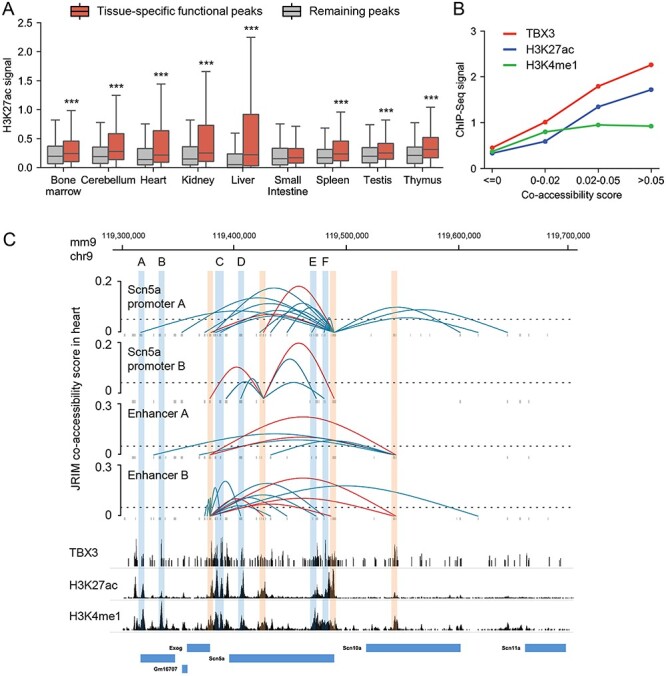
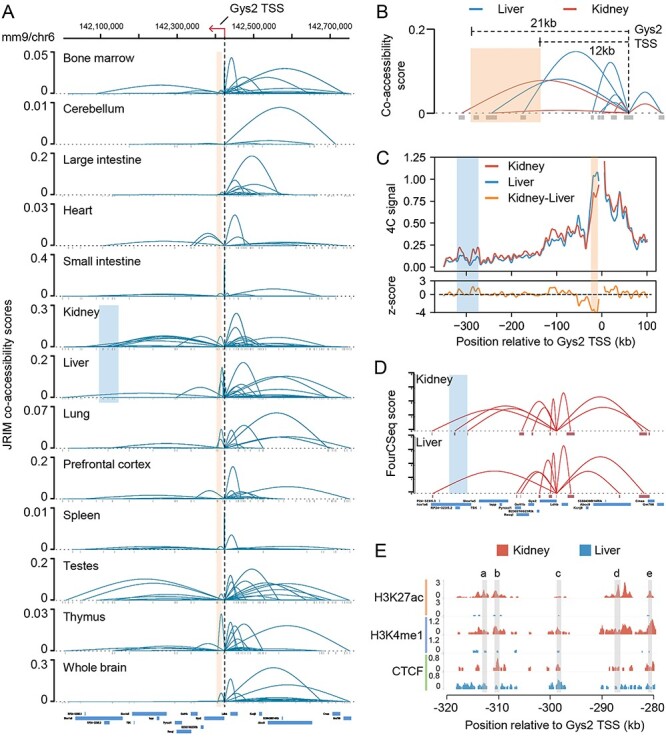
The rapid accumulation of single-cell chromatin accessibility data offers a unique opportunity to investigate common and specific regulatory mechanisms across different cell types. However, existing methods for cis-regulatory network reconstruction using single-cell chromatin accessibility data were only designed for cells belonging to one cell type, and resulting networks may be incomparable directly due to diverse cell numbers of different cell types. Here, we adopt a computational method to jointly reconstruct cis-regulatory interaction maps (JRIM) of multiple cell populations based on patterns of co-accessibility in single-cell data. We applied JRIM to explore common and specific regulatory interactions across multiple tissues from single-cell ATAC-seq dataset containing ~80 000 cells across 13 mouse tissues. Reconstructed common interactions among 13 tissues indeed relate to basic biological functions, and individual cis-regulatory networks show strong tissue specificity and functional relevance. More importantly, tissue-specific regulatory interactions are mediated by coordination of histone modifications and tissue-related TFs, and many of them may reveal novel regulatory mechanisms.
Keywords: cis-regulatory interaction networks; Gaussian graphical LASSO; single-cell ATAC-seq.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet 2012;13:59–69. - PubMed
-
- Yang X, Shao X, Gao L, et al. . Systematic DNA methylation analysis of multiple cell lines reveals common and specific patterns within and across tissues of origin. Hum Mol Genet 2015;24:4374–84. - PubMed
