

Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History
- PMID: 32580384
- PMCID: PMC7352894
- DOI: 10.3390/ijms21124435
Molecular Epidemiology of B3 and D8 Measles Viruses through Hemagglutinin Phylogenetic History
Abstract
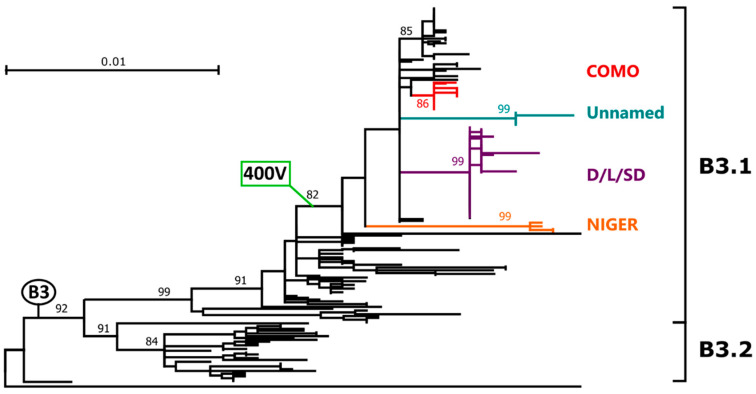
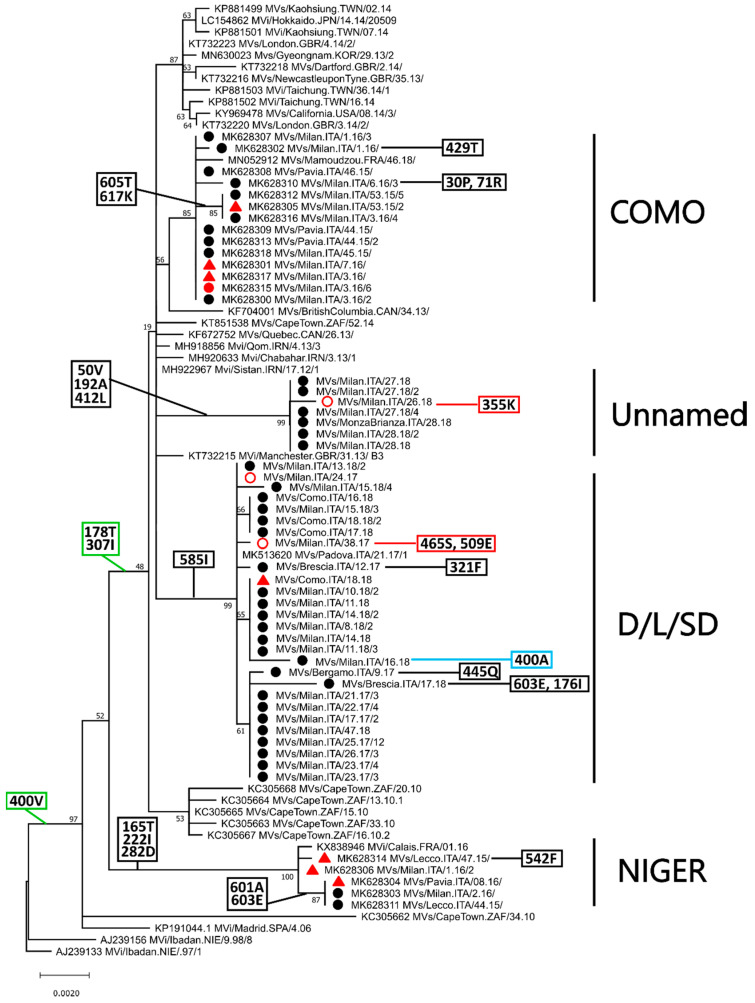
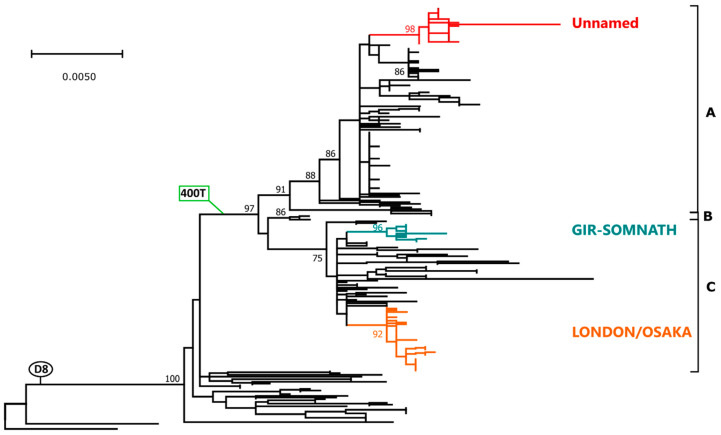
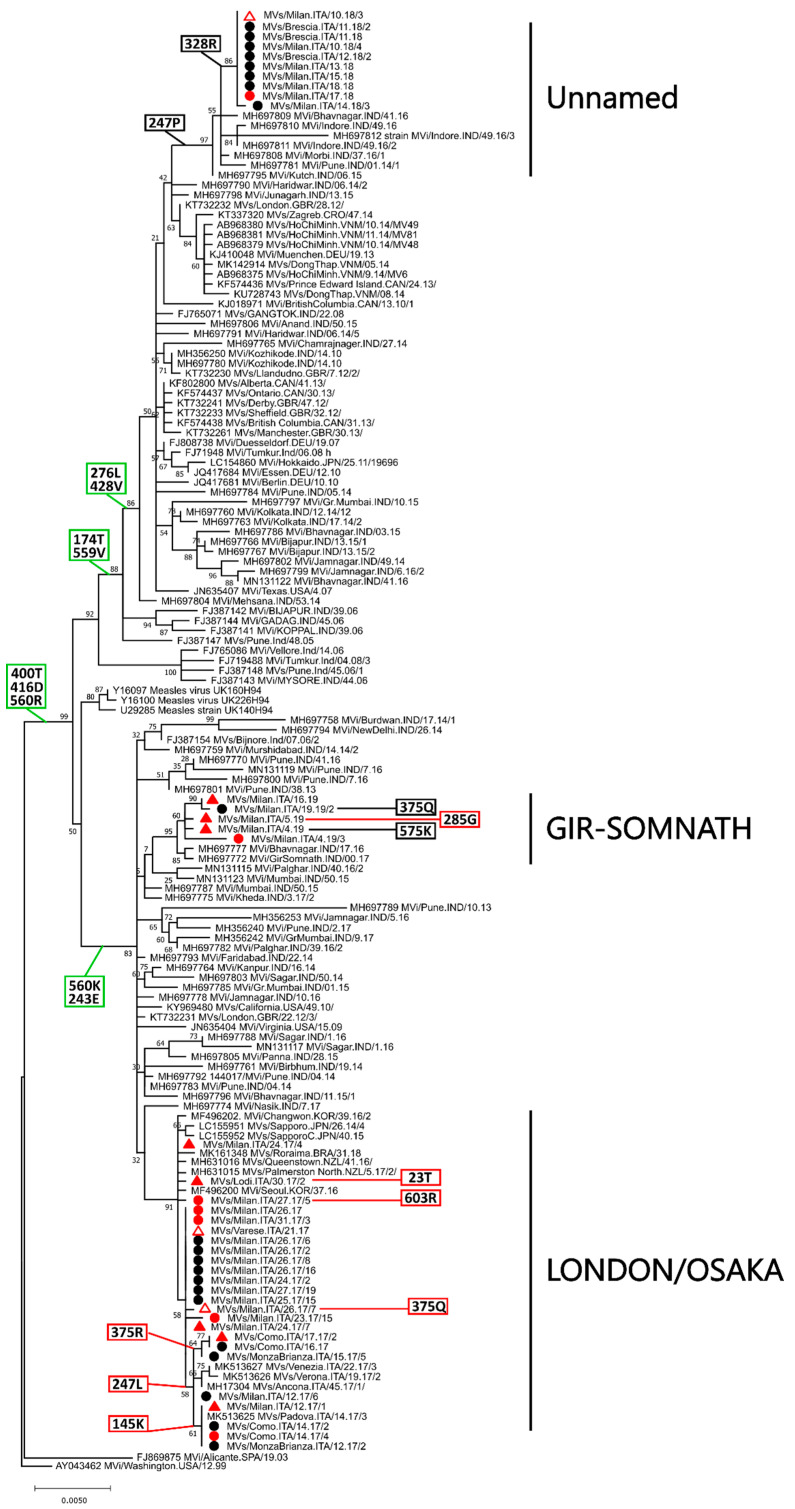
Of the 24 known measles genotypes, only D8 and B3 are responsible for outbreaks in the last years in Europe, Asia, and America. In this study the H gene of 92 strains circulating between 2015 and 2019 in Lombardy, Northern Italy, and 1273 H sequences available in GenBank were analyzed in order to evaluate the genetic variability and to assess the conservation of the immunodominant sites. Overall, in Lombardy we observed the presence of four different B3 and three different D8 clusters, each one of them including sequences derived from viruses found in both vaccinated and unvaccinated subjects. Worldwide, the residue 400 within the H protein, a position located within the main immune epitope, is mutated in all circulating strains that belong to the two globally endemic genotypes, B3 and D8. Our data demonstrate the usefulness of measles virus (MV) H gene sequencing. Indeed, the monitoring the H protein epitopes of circulating strains could be included in the measles laboratory surveillance activities in order to improve and optimize strategies for measles control, as countries go towards elimination phase.
Keywords: H gene; genotype B3; genotype D8; hemagglutinin; immunodominant epitopes; measles surveillance strategies; measles virus.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Role of Sequencing the Measles Virus Hemagglutinin Gene and Hypervariable Region in the Measles Outbreak Investigations in Sweden During 2013-2014.J Infect Dis. 2016 Feb 15;213(4):592-9. doi: 10.1093/infdis/jiv434. Epub 2015 Sep 7. J Infect Dis. 2016. PMID: 26347574
-
Measles re-emergence in Northern Italy: Pathways of measles virus genotype D8, 2013-2014.Infect Genet Evol. 2017 Mar;48:120-126. doi: 10.1016/j.meegid.2016.12.013. Epub 2016 Dec 15. Infect Genet Evol. 2017. PMID: 27989661
-
Towards measles elimination: Phylogenetic analysis of measles viruses in Turkey (2012-2013) and identification of genotype D8.J Med Virol. 2016 Nov;88(11):1867-73. doi: 10.1002/jmv.24548. Epub 2016 Apr 26. J Med Virol. 2016. PMID: 27089242
-
Molecular epidemiology of measles virus.Curr Top Microbiol Immunol. 2009;330:129-50. doi: 10.1007/978-3-540-70617-5_7. Curr Top Microbiol Immunol. 2009. PMID: 19203108 Review.
-
Global genetic diversity of measles virus (Paramyxoviridae: Morbillivirus: Morbillivirus hominis): historical aspects and current state.Vopr Virusol. 2023 Nov 7;68(5):361-371. doi: 10.36233/0507-4088-187. Vopr Virusol. 2023. PMID: 38156571 Review.
Cited by
-
Breakthrough Infections: A Challenge towards Measles Elimination?Microorganisms. 2022 Aug 4;10(8):1567. doi: 10.3390/microorganisms10081567. Microorganisms. 2022. PMID: 36013985 Free PMC article. Review.
-
Characterization of Vaccine Breakthrough Cases during Measles Outbreaks in Milan and Surrounding Areas, Italy, 2017-2021.Viruses. 2022 May 17;14(5):1068. doi: 10.3390/v14051068. Viruses. 2022. PMID: 35632809 Free PMC article.
-
Genetic Characterizations and Molecular Evolution of the Measles Virus Genotype B3's Hemagglutinin (H) Gene in the Elimination Era.Viruses. 2021 Sep 30;13(10):1970. doi: 10.3390/v13101970. Viruses. 2021. PMID: 34696400 Free PMC article.
-
Serosurvey of anti-rubella and anti-measles IgG antibodies in young females in Jahrom, southern west Iran in 2012: A review of literature of the serological profile in Iran.J Gen Fam Med. 2024 Feb 25;25(2):95-101. doi: 10.1002/jgf2.677. eCollection 2024 Mar. J Gen Fam Med. 2024. PMID: 38481743 Free PMC article.
-
Impact of genotypic variability of measles virus T-cell epitopes on vaccine-induced T-cell immunity.NPJ Vaccines. 2025 Feb 20;10(1):36. doi: 10.1038/s41541-025-01088-y. NPJ Vaccines. 2025. PMID: 39979288 Free PMC article.
References
-
- Patel M., Lee A.D., Clemmons N.S., Redd S.B., Poser S., Blog D., Zucker J.R., Leung J., Link-Gelles R., Pham H., et al. National Update on Measles Cases and Outbreaks-United States, January 1-October 1, 2019. MMWR Morb. Mortal. Wkly. Rep. 2019;68:893–896. doi: 10.15585/mmwr.mm6840e2. - DOI - PMC - PubMed
