

Initial dose of oncolytic myxoma virus programs durable antitumor immunity independent of in vivo viral replication
- PMID: 32581062
- PMCID: PMC7319776
- DOI: 10.1136/jitc-2020-000804
Initial dose of oncolytic myxoma virus programs durable antitumor immunity independent of in vivo viral replication
Abstract
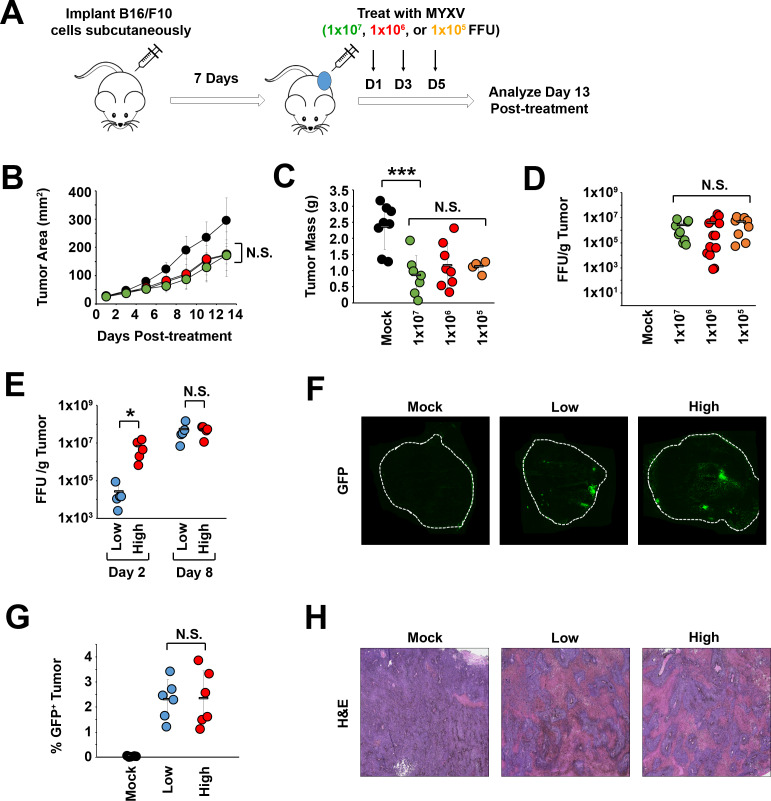
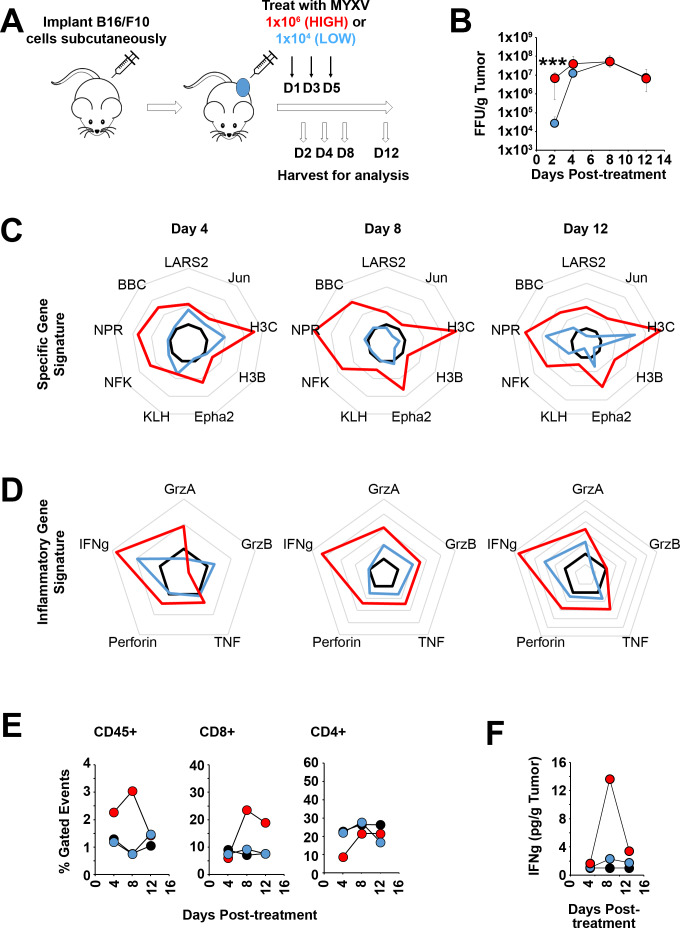
Background: Oncolytic therapy uses live-replicating viruses to improve the immunological status of treated tumors. Critically, while these viruses are known to self-amplify in vivo, clinical oncolytic therapies still appear to display a strong dose dependence and the mechanisms mediating this dose dependence are not well understood.
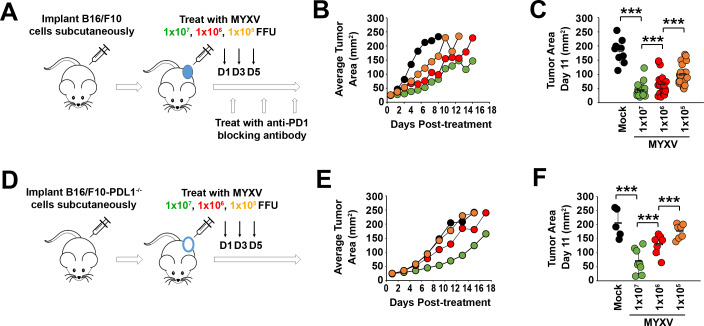
Methods: To explore this apparent contradiction, we investigated how the initial dose of oncolytic myxoma virus affected the subsequent ability of treatment to alter the immunological status of tumors as well as synergize with programmed cell death protein 1 (PD1) blockade.
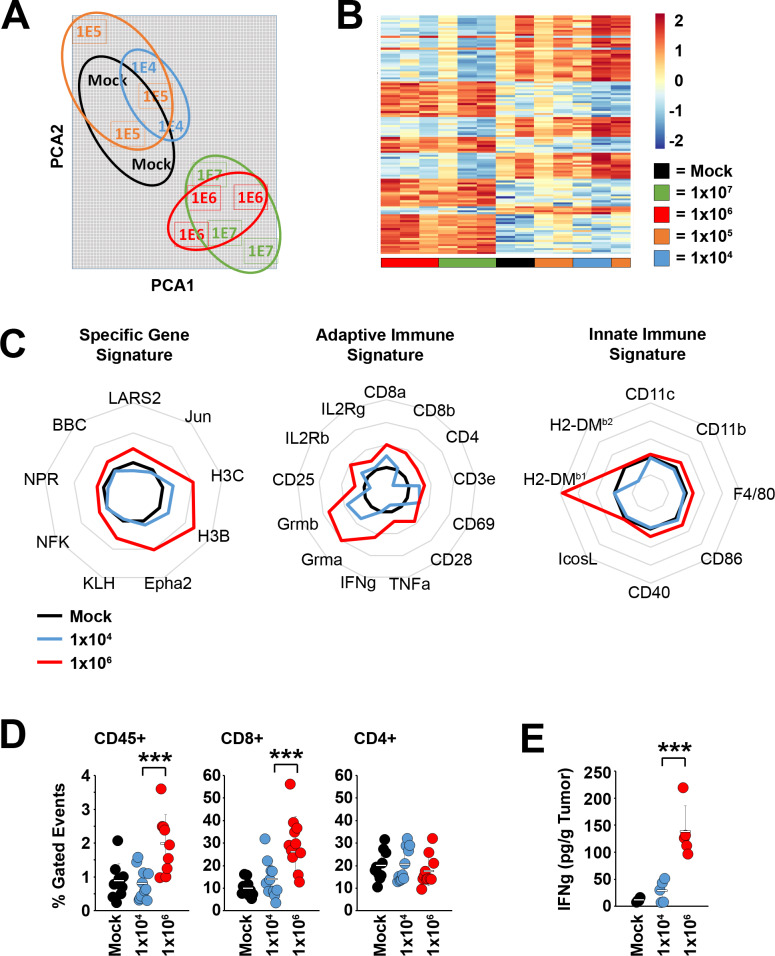
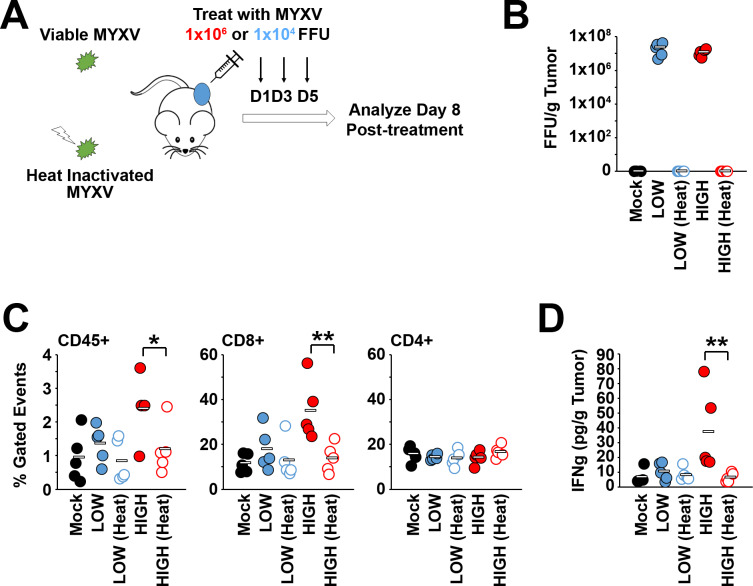
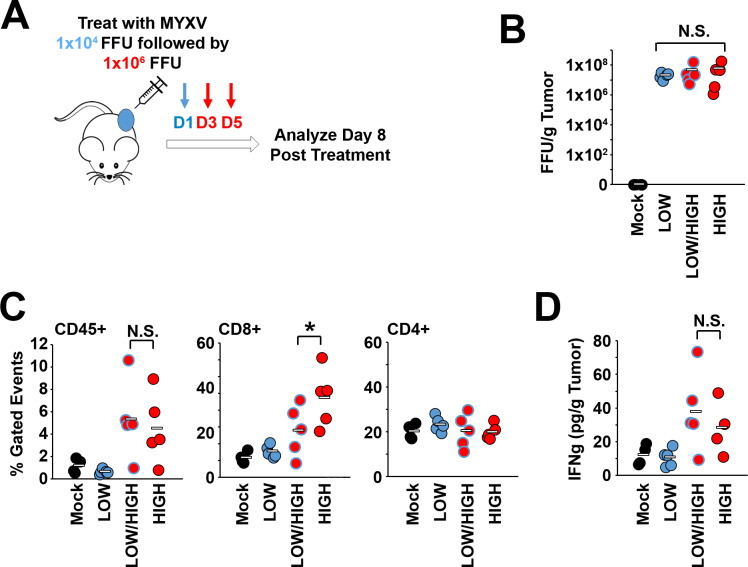
Results: Our results indicate that, due to viral self-amplification in vivo, the overall load of myxoma virus rapidly normalizes within treated tumors despite up to 3-log differences in inoculating dose. Because of this, therapeutic efficacy in the absence of checkpoint blockade is largely dose independent. Despite this rapid normalization, however, treatment with high or low doses of myxoma virus induces distinct immunological changes within treated tumors. Critically, these changes appear to be durably programmed based on the initial oncolytic dose with low-dose treatment failing to induce immunological improvements despite rapidly achieving equivalent viral burdens. Finally, due to the distinct immunological profiles induced by high and low myxoma virus doses, oncolytic efficacy resulting from combination with PD1 blockade therapy displays a strong dose dependence.
Conclusions: Taken together, these data suggest that the ability of oncolytic myxoma virus to immunologically reprogram treated tumors is dependent on initial viral dose. Additionally, this work could provide a possible mechanistic explanation for clinical results observed with other oncolytic viruses.
Keywords: oncolytic virotherapy; oncolytic viruses.
© Author(s) (or their employer(s)) 2020. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Tomioka N, Azuma M, Ikarashi M, et al. The therapeutic candidate for immune checkpoint inhibitors elucidated by the status of tumor-infiltrating lymphocytes (TILs) and programmed death ligand 1 (PD-L1) expression in triple negative breast cancer (TNBC). Breast Cancer 2018;25:34–42. 10.1007/s12282-017-0781-0 - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials