

Pervasive lesion segregation shapes cancer genome evolution
- PMID: 32581361
- PMCID: PMC7116693
- DOI: 10.1038/s41586-020-2435-1
Pervasive lesion segregation shapes cancer genome evolution
Abstract
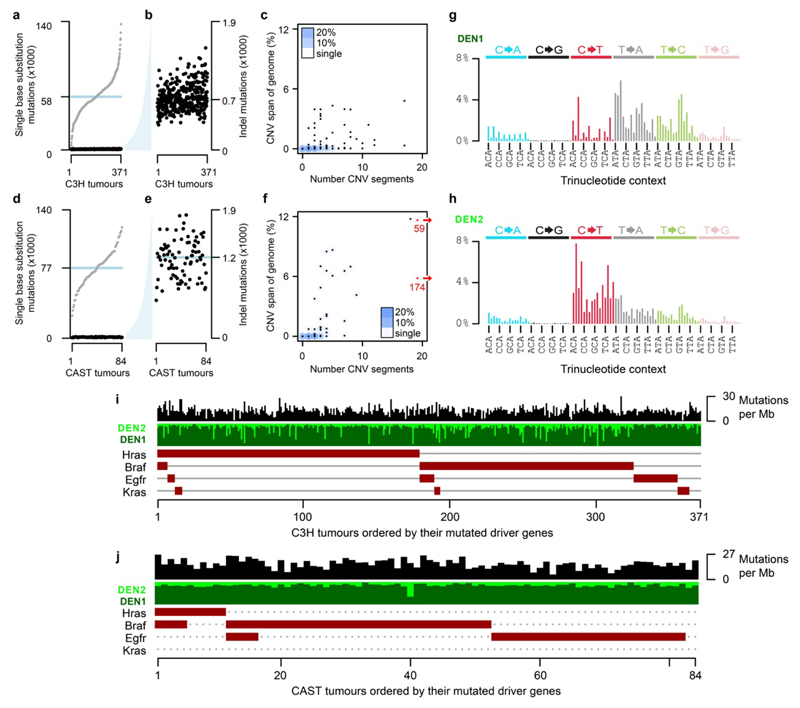
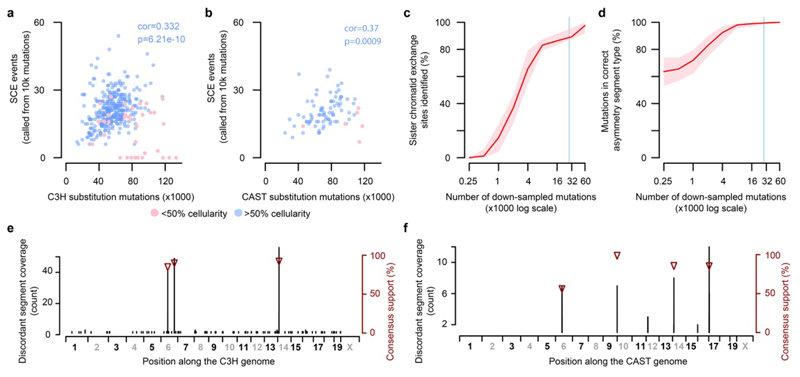
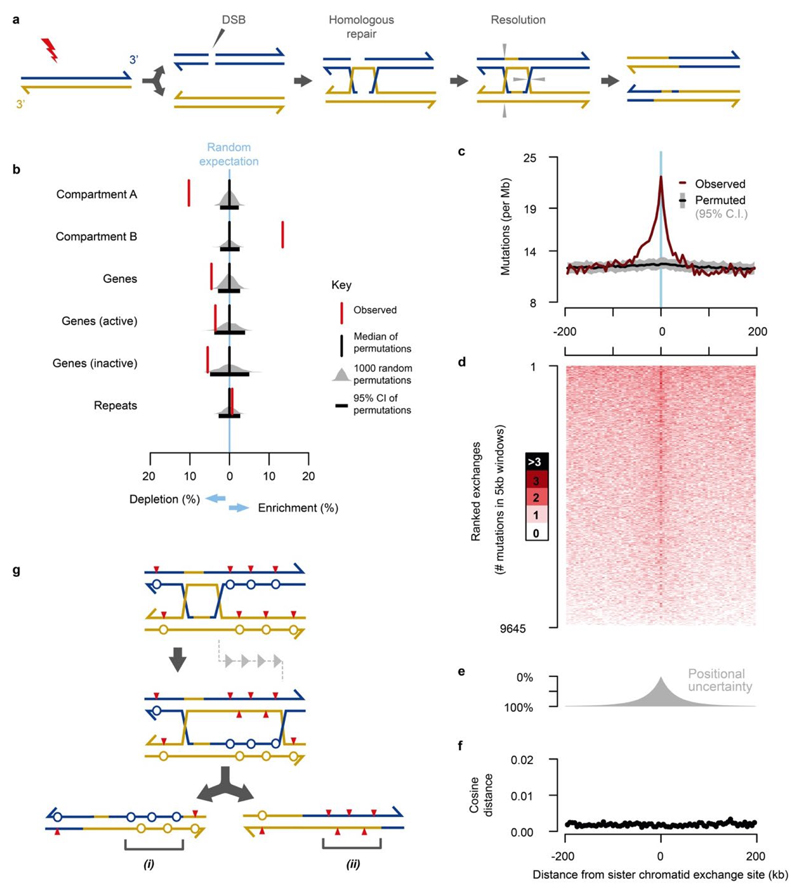
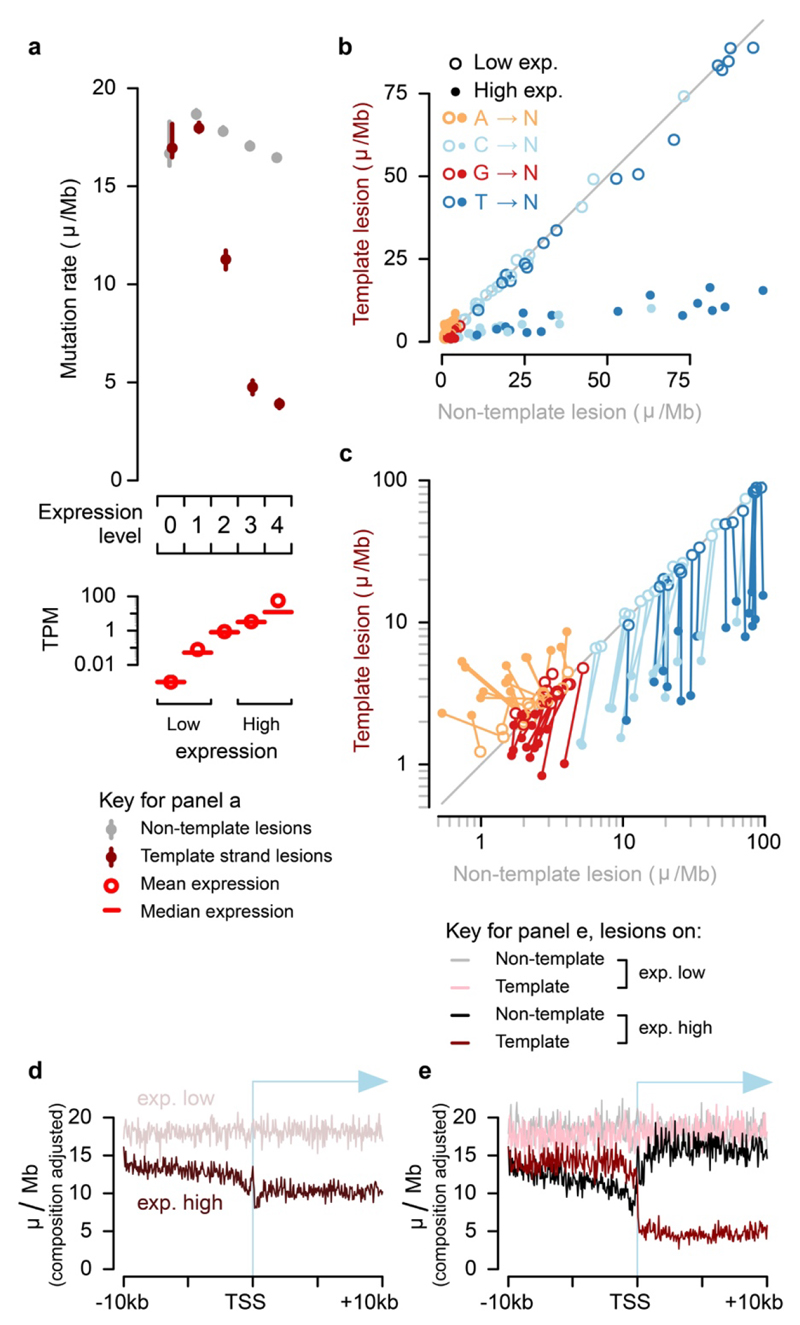
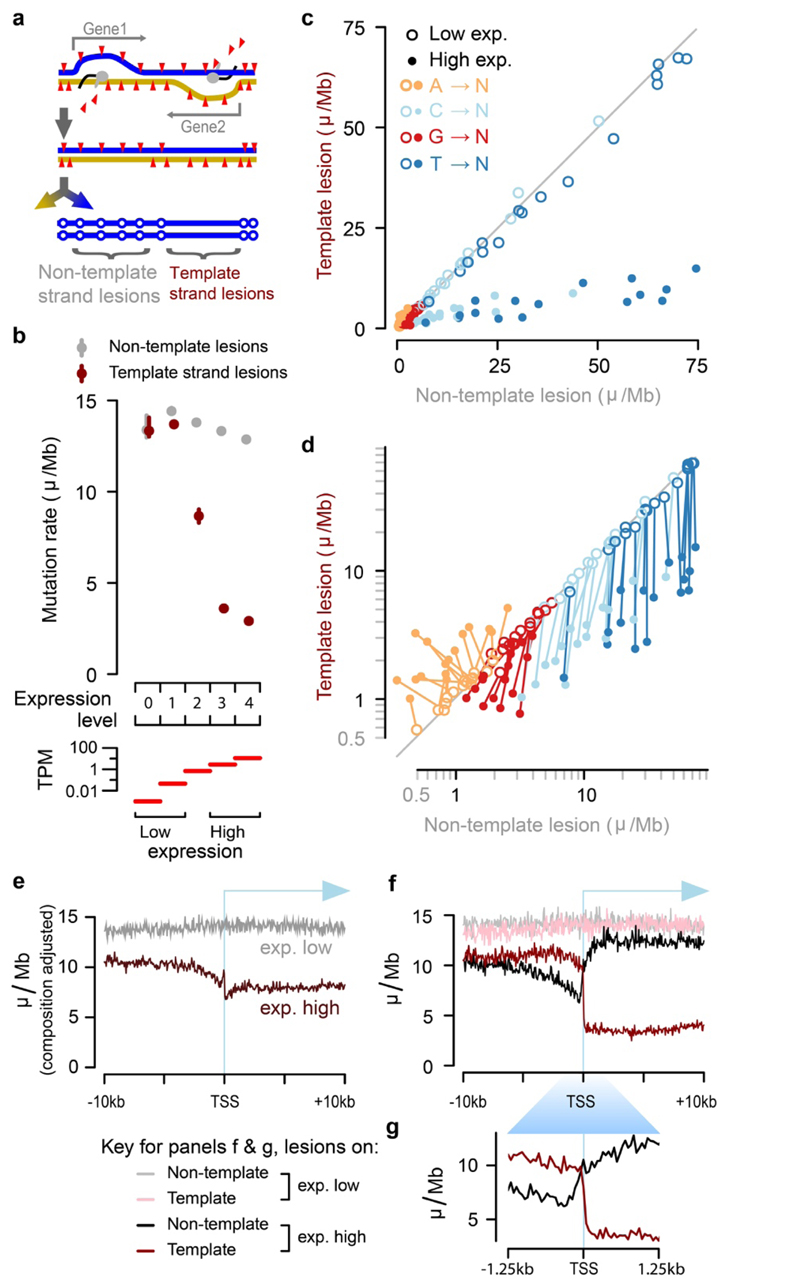
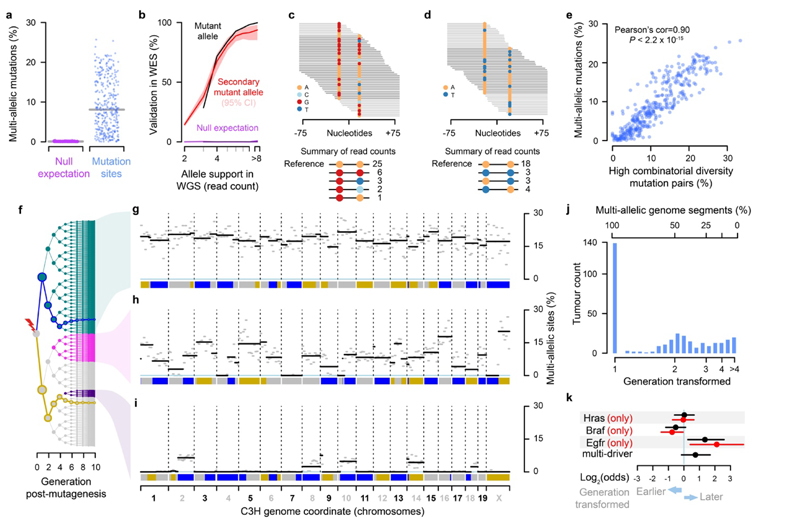
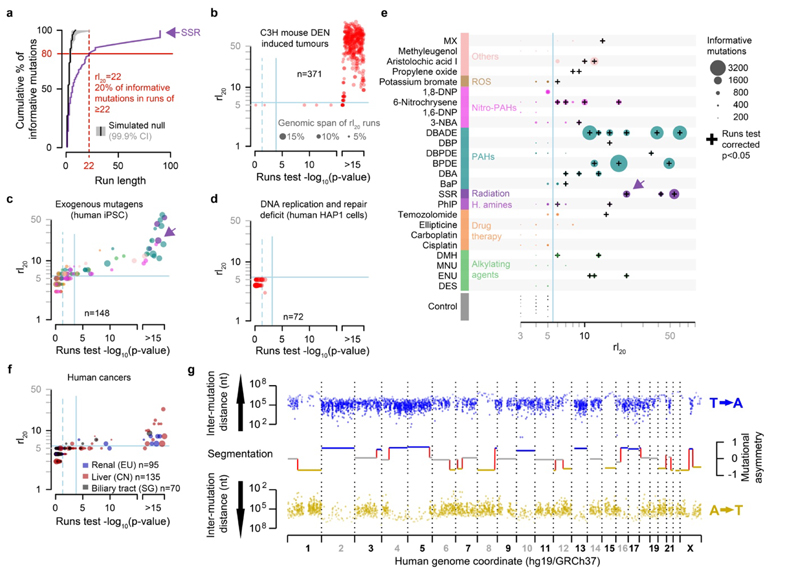
Cancers arise through the acquisition of oncogenic mutations and grow by clonal expansion1,2. Here we reveal that most mutagenic DNA lesions are not resolved into a mutated DNA base pair within a single cell cycle. Instead, DNA lesions segregate, unrepaired, into daughter cells for multiple cell generations, resulting in the chromosome-scale phasing of subsequent mutations. We characterize this process in mutagen-induced mouse liver tumours and show that DNA replication across persisting lesions can produce multiple alternative alleles in successive cell divisions, thereby generating both multiallelic and combinatorial genetic diversity. The phasing of lesions enables accurate measurement of strand-biased repair processes, quantification of oncogenic selection and fine mapping of sister-chromatid-exchange events. Finally, we demonstrate that lesion segregation is a unifying property of exogenous mutagens, including UV light and chemotherapy agents in human cells and tumours, which has profound implications for the evolution and adaptation of cancer genomes.
Conflict of interest statement
P.F. is a member of the Scientific Advisory Boards of Fabric Genomics, Inc., and Eagle Genomics, Ltd.
Figures
Comment in
-
Strands of evidence about cancer evolution.Nature. 2020 Jul;583(7815):207-209. doi: 10.1038/d41586-020-01815-6. Nature. 2020. PMID: 32620881 No abstract available.
-
Strands of evolution.Nat Rev Cancer. 2020 Sep;20(9):483. doi: 10.1038/s41568-020-0292-8. Nat Rev Cancer. 2020. PMID: 32669634 No abstract available.
References
-
- Turajlic S, Sottoriva A, Graham T, Swanton C. Resolving genetic heterogeneity in cancer. Nat Rev Genet. 2019;20:404–416. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- WT108749/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- MC_UU_00007/16/MRC_/Medical Research Council/United Kingdom
- MC_UU_00009/2/MRC_/Medical Research Council/United Kingdom
- MC_UU_00007/11/MRC_/Medical Research Council/United Kingdom
- WT202878/B/16/Z/WT_/Wellcome Trust/United Kingdom
- 20412/CRUK_/Cancer Research UK/United Kingdom
- 22398/CRUK_/Cancer Research UK/United Kingdom
- WT106563/Z/14/A/WT_/Wellcome Trust/United Kingdom
- 202878/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- A20412/CRUK_/Cancer Research UK/United Kingdom
- 615584/ERC_/European Research Council/International
- A22398/CRUK_/Cancer Research UK/United Kingdom
- MR/R026017/1/MRC_/Medical Research Council/United Kingdom
- 202878/WT_/Wellcome Trust/United Kingdom
- 106563/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
