

Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data
- PMID: 32584579
- PMCID: PMC7490796
- DOI: 10.1021/acs.jproteome.0c00332
Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data
Abstract
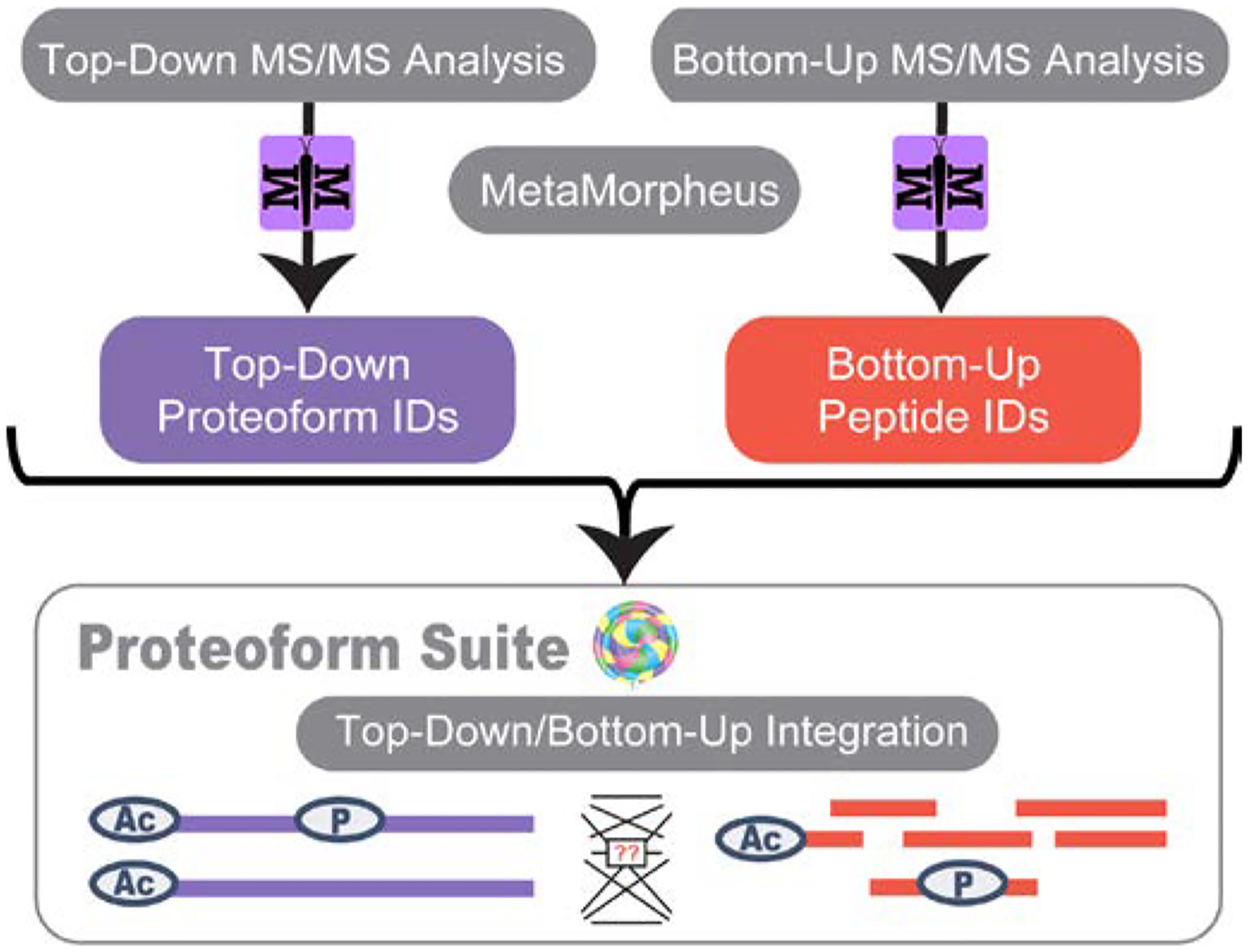
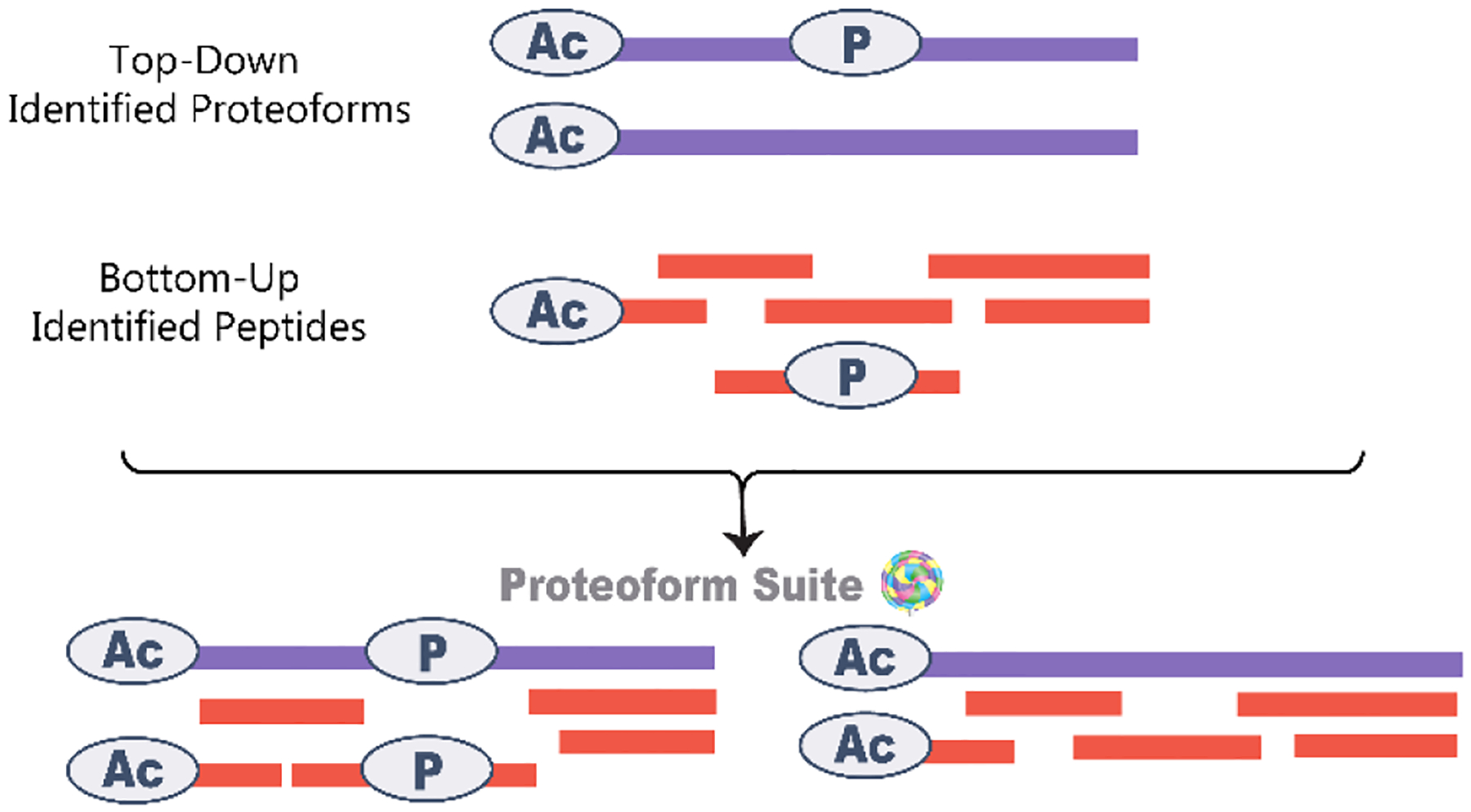
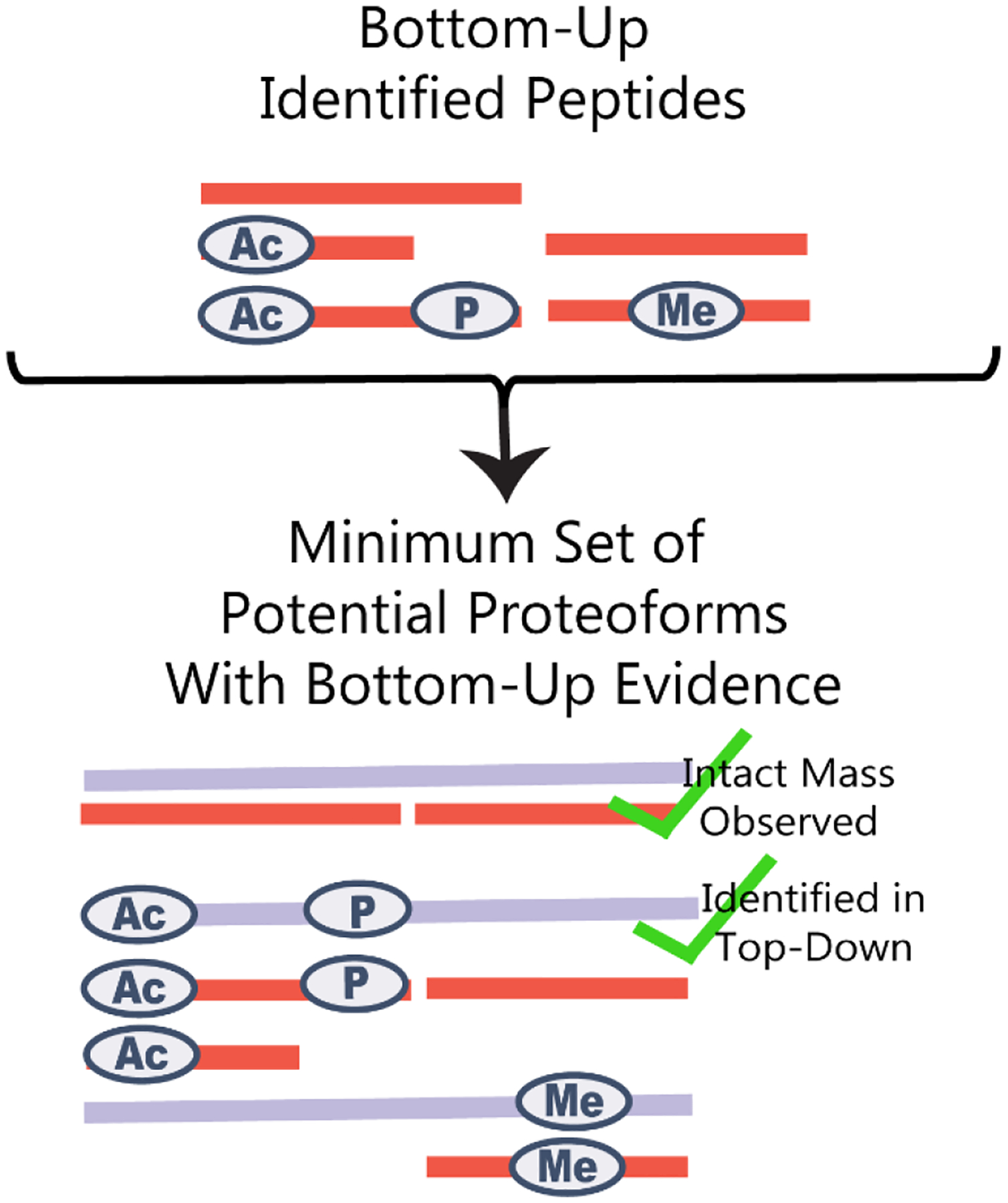
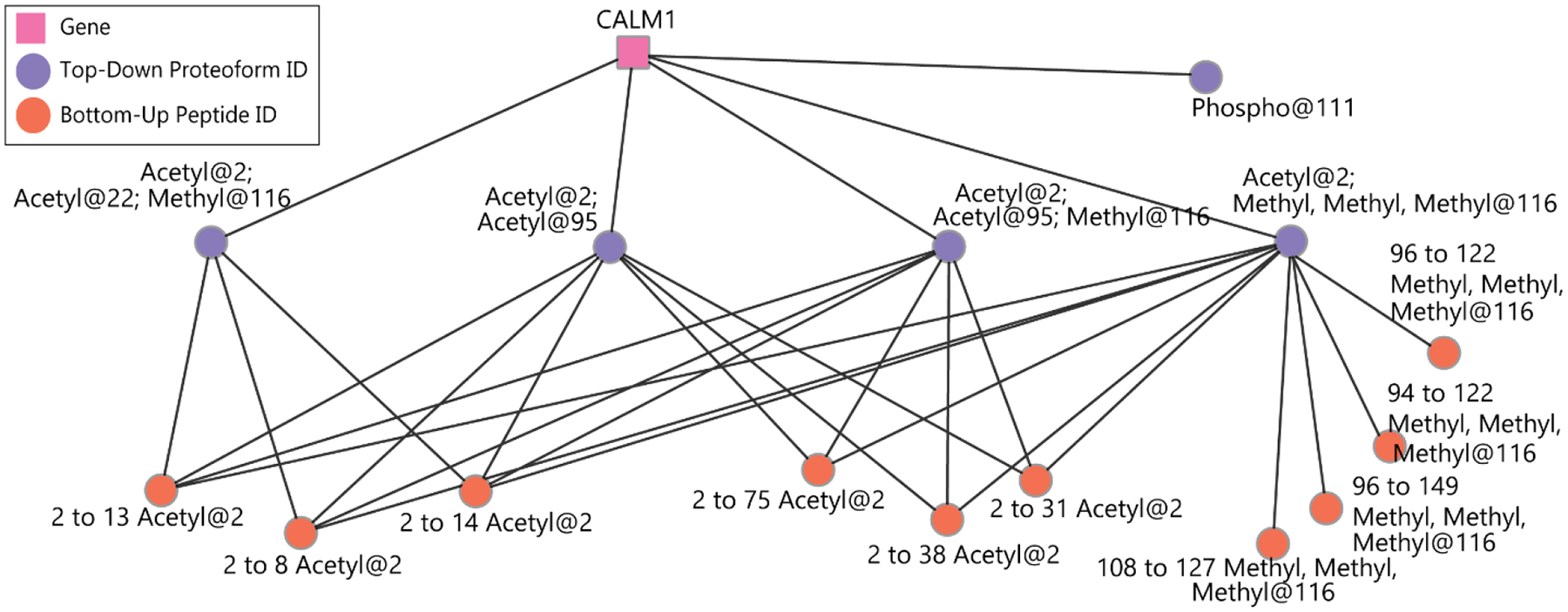
Cellular functions are performed by a vast and diverse set of proteoforms. Proteoforms are the specific forms of proteins produced as a result of genetic variations, RNA splicing, and post-translational modifications (PTMs). Top-down mass spectrometric analysis of intact proteins enables proteoform identification, including proteoforms derived from sequence cleavage events or harboring multiple PTMs. In contrast, bottom-up proteomics identifies peptides, which necessitates protein inference and does not yield proteoform identifications. We seek here to exploit the synergies between these two data types to improve the quality and depth of the overall proteomic analysis. To this end, we automated the large-scale integration of results from multiprotease bottom-up and top-down analyses in the software program Proteoform Suite and applied it to the analysis of proteoforms from the human Jurkat T lymphocyte cell line. We implemented the recently developed proteoform-level classification scheme for top-down tandem mass spectrometry (MS/MS) identifications in Proteoform Suite, which enables users to observe the level and type of ambiguity for each proteoform identification, including which of the ambiguous proteoform identifications are supported by bottom-up-level evidence. We used Proteoform Suite to find instances where top-down identifications aid in protein inference from bottom-up analysis and conversely where bottom-up peptide identifications aid in proteoform PTM localization. We also show the use of bottom-up data to infer proteoform candidates potentially present in the sample, allowing confirmation of such proteoform candidates by intact-mass analysis of MS1 spectra. The implementation of these capabilities in the freely available software program Proteoform Suite enables users to integrate large-scale top-down and bottom-up data sets and to utilize the synergies between them to improve and extend the proteomic analysis.
Keywords: bottom-up proteomics; post-translational modification; protein inference; proteoforms; software; top-down proteomics.
Conflict of interest statement
The authors declare no competing financial interests.
Figures




References
-
- Yang X; Coulombe-Huntington J; Kang S; Sheynkman GM; Hao T; Richardson A; Sun S; Yang F; Shen YA; Murray RR; Spirohn K; Begg BE; Duran-Frigola M; MacWilliams A; Pevzner SJ; Zhong Q; Trigg SA; Tam S; Ghamsari L; Sahni N; Yi S; Rodriguez MD; Balcha D; Tan G; Costanzo M; Andrews B; Boone C; Zhou XJ; Salehi-Ashtiani K; Charloteaux B; Chen AA; Calderwood MA; Aloy P; Roth FP; Hill DE; Iakoucheva LM; Xia Y; Vidal M, Widespread Expansion of Protein Interaction Capabilities by Alternative Splicing. Cell 2016, 164, (4), 805–17. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
