

Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes
- PMID: 32584970
- PMCID: PMC7498364
- DOI: 10.1182/blood.2020005289
Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes
Abstract
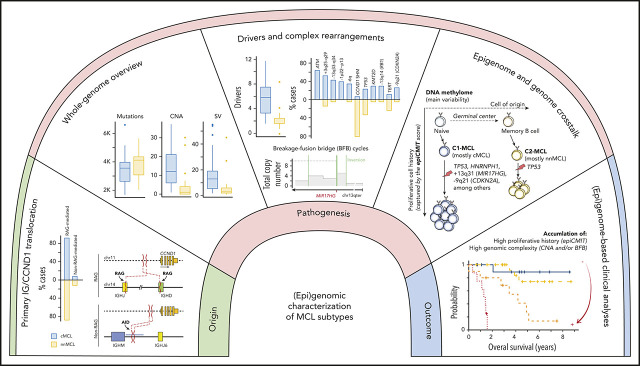
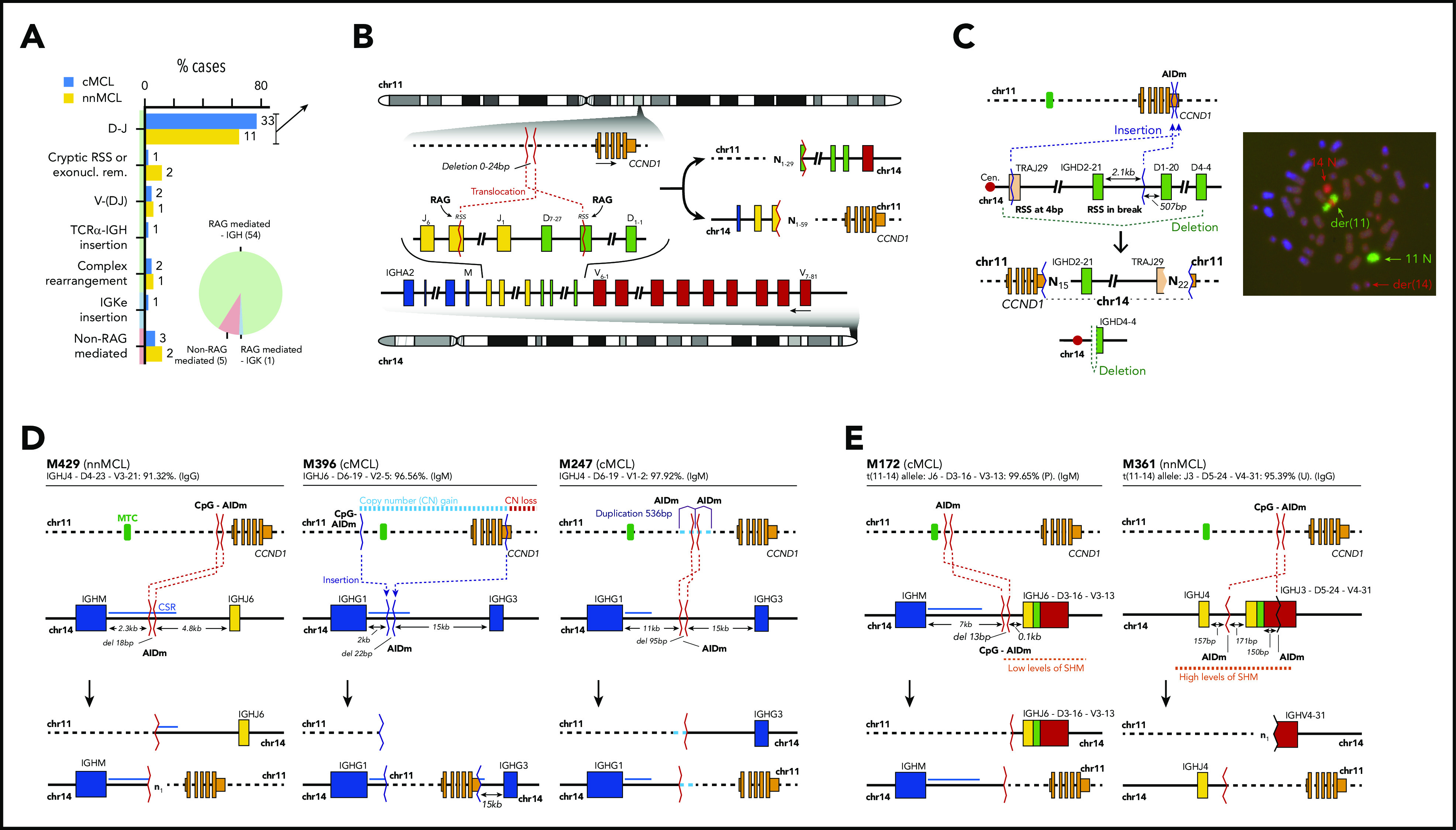
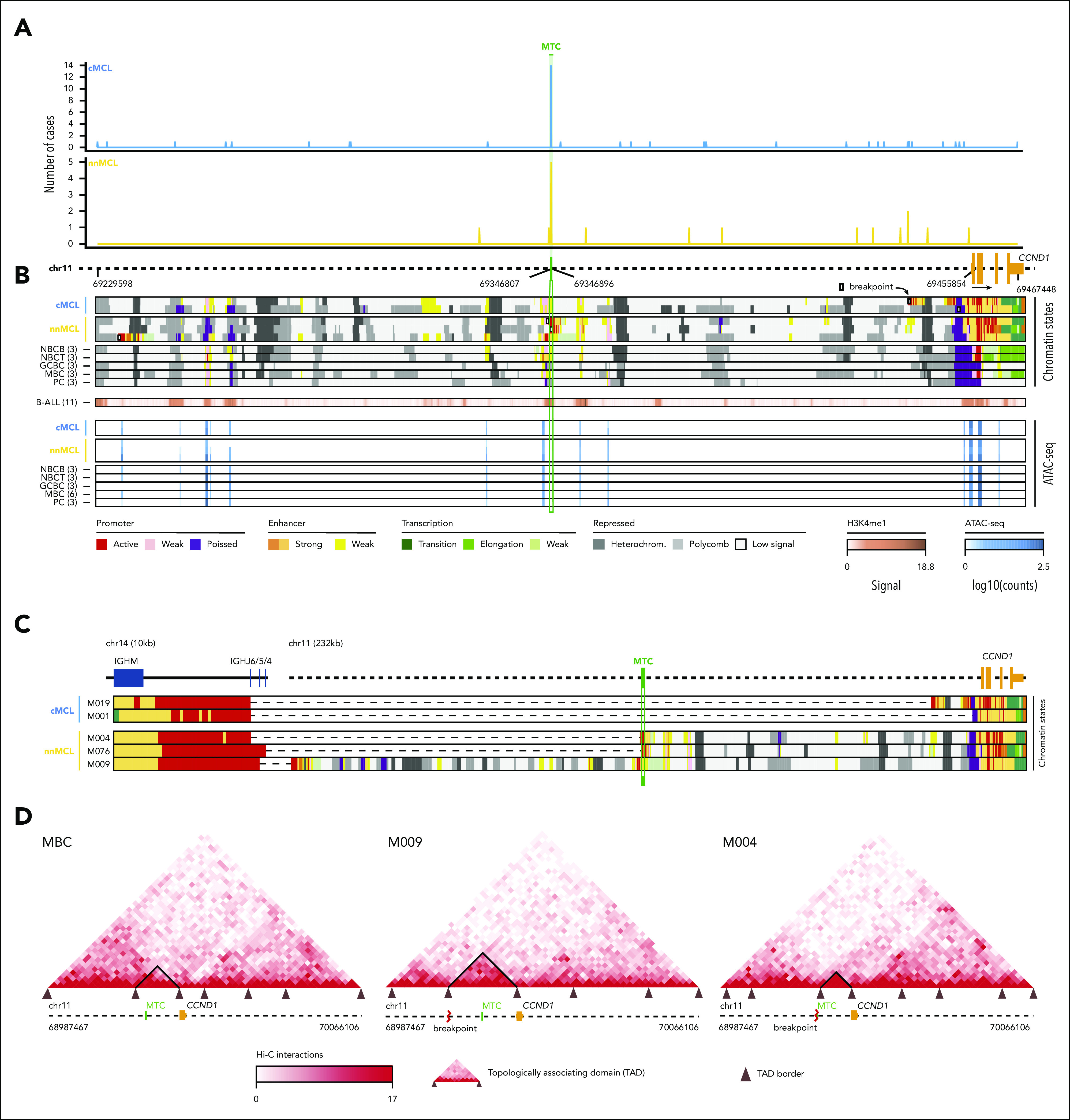
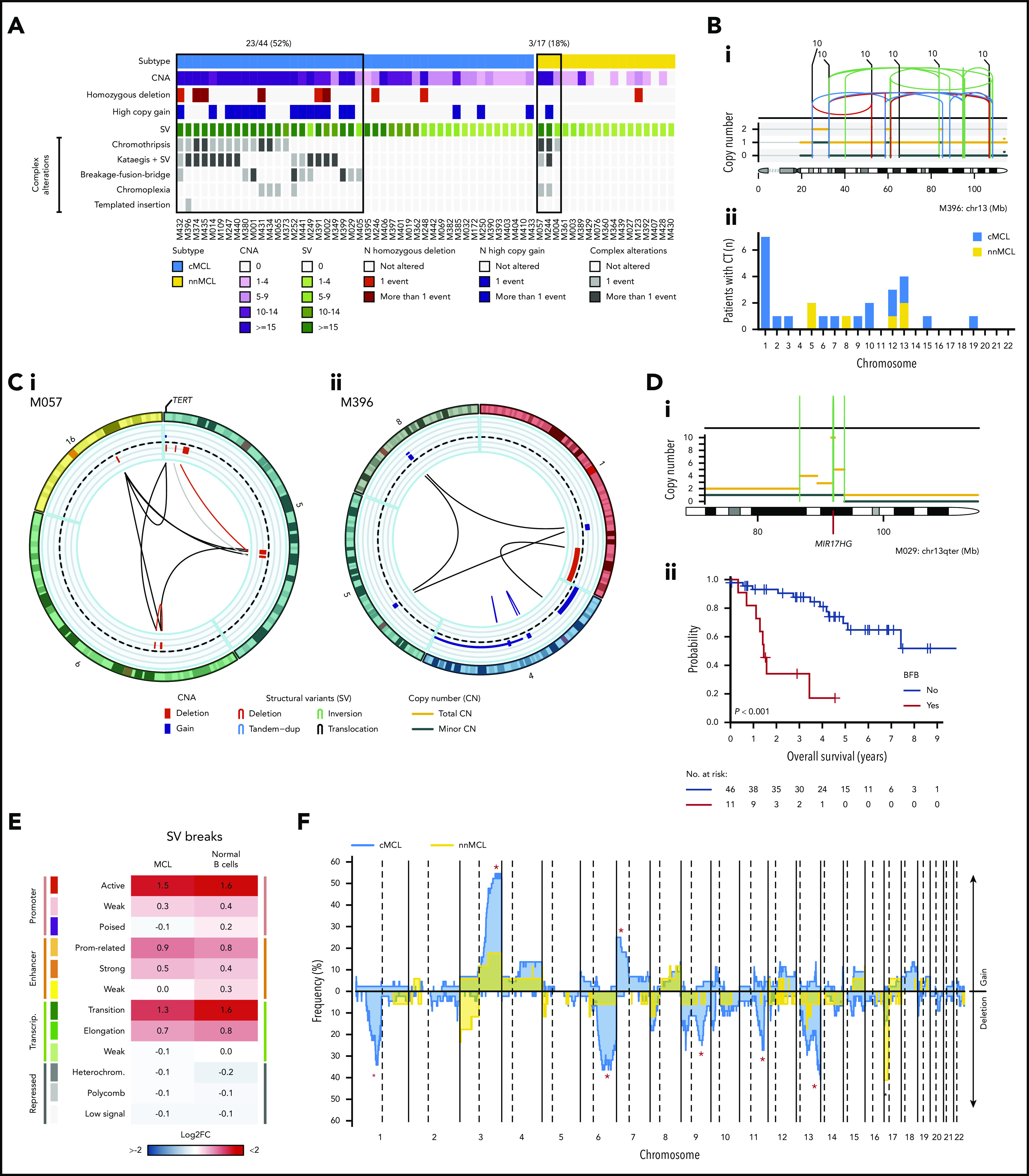
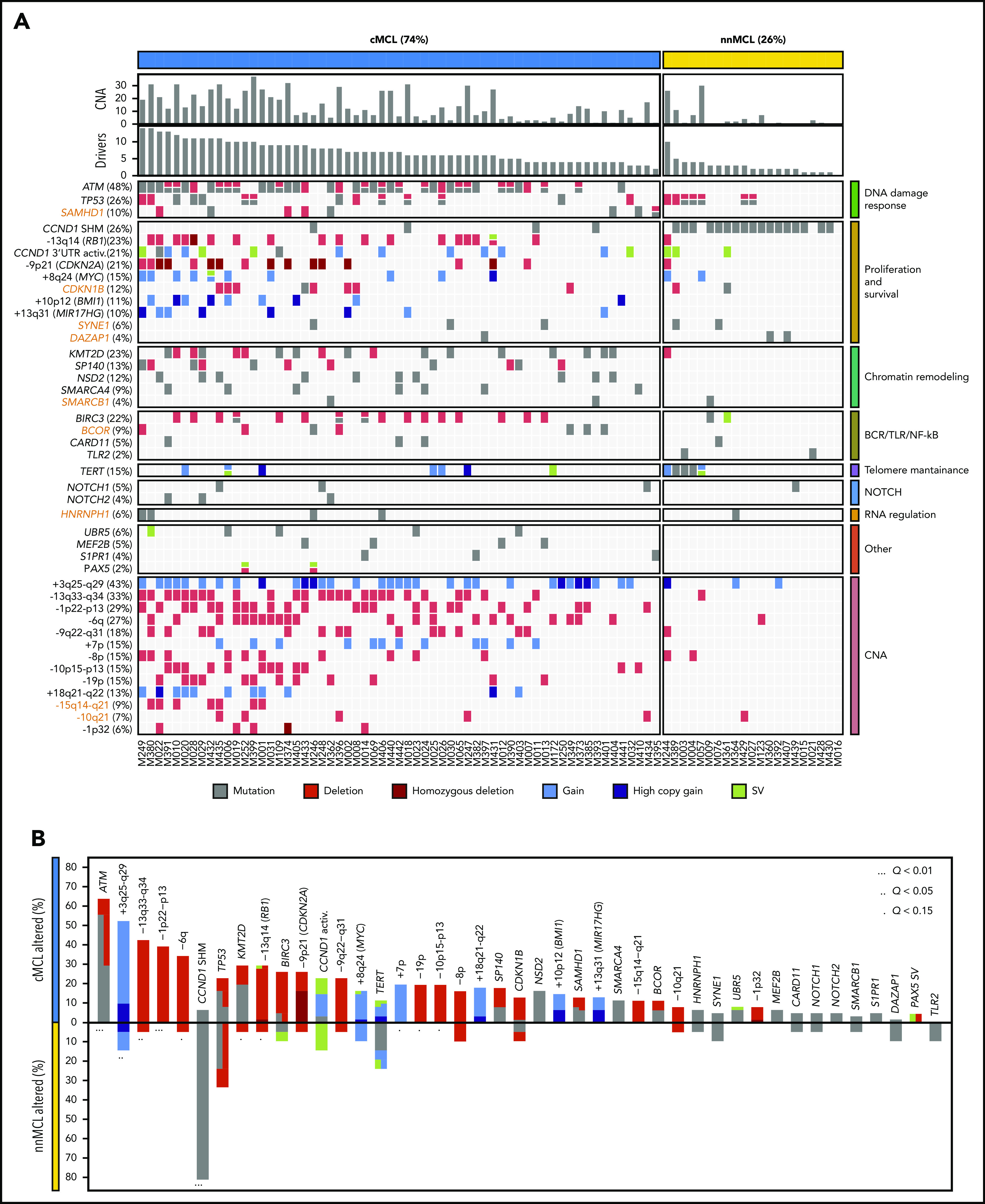
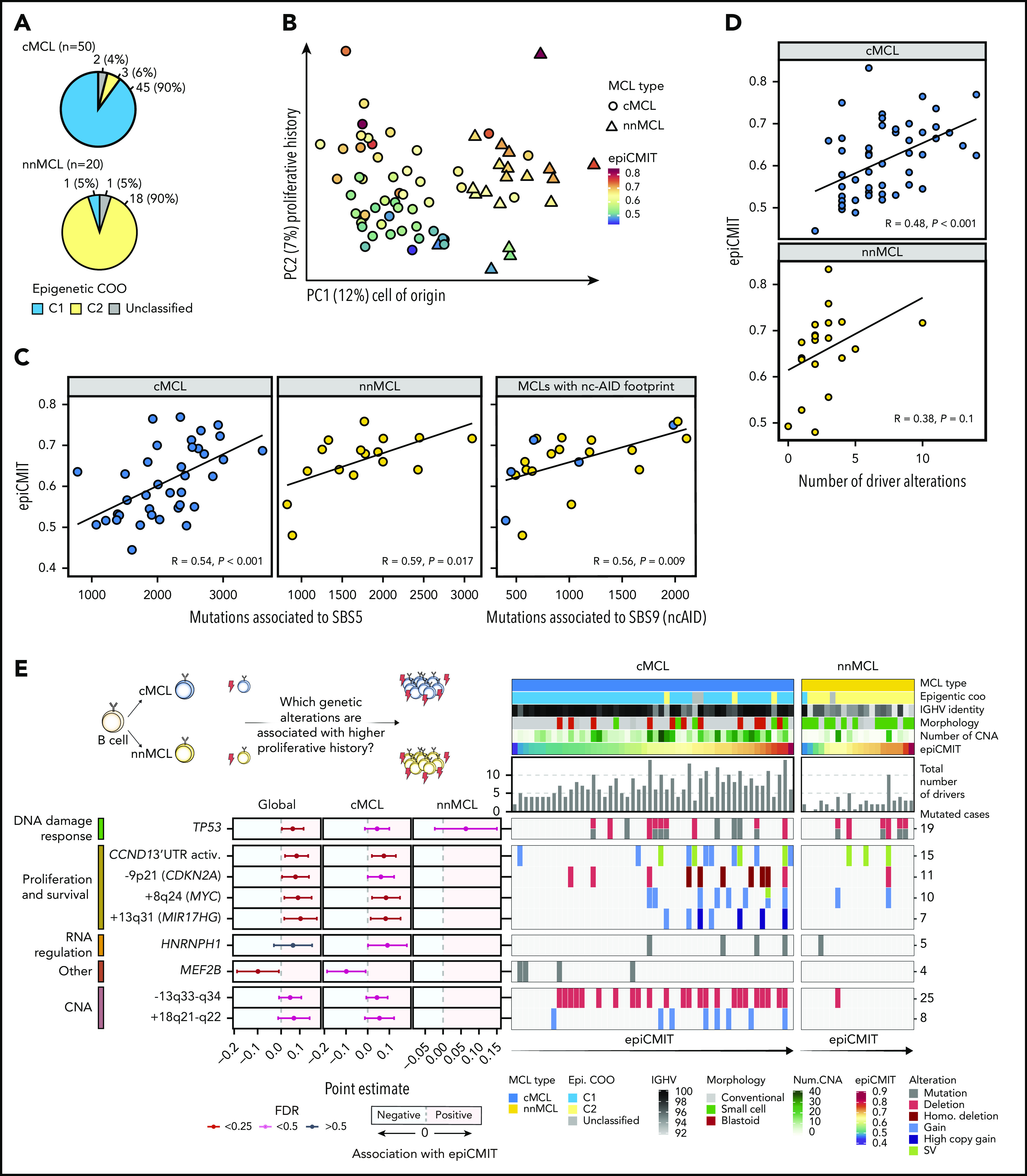
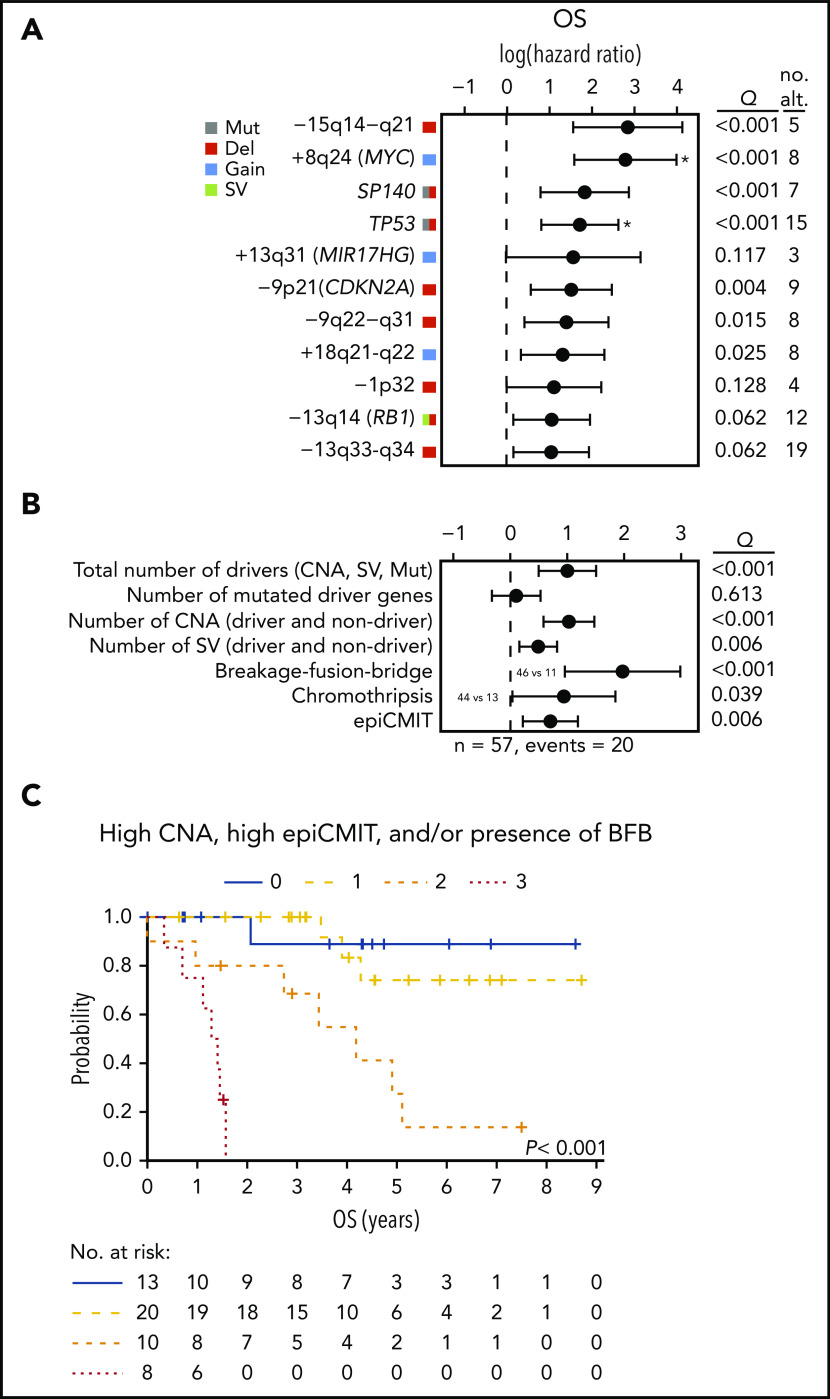
Mantle cell lymphoma (MCL) is a mature B-cell neoplasm initially driven by CCND1 rearrangement with 2 molecular subtypes, conventional MCL (cMCL) and leukemic non-nodal MCL (nnMCL), that differ in their clinicobiological behavior. To identify the genetic and epigenetic alterations determining this diversity, we used whole-genome (n = 61) and exome (n = 21) sequencing (74% cMCL, 26% nnMCL) combined with transcriptome and DNA methylation profiles in the context of 5 MCL reference epigenomes. We identified that open and active chromatin at the major translocation cluster locus might facilitate the t(11;14)(q13;32), which modifies the 3-dimensional structure of the involved regions. This translocation is mainly acquired in precursor B cells mediated by recombination-activating genes in both MCL subtypes, whereas in 8% of cases the translocation occurs in mature B cells mediated by activation-induced cytidine deaminase. We identified novel recurrent MCL drivers, including CDKN1B, SAMHD1, BCOR, SYNE1, HNRNPH1, SMARCB1, and DAZAP1. Complex structural alterations emerge as a relevant early oncogenic mechanism in MCL, targeting key driver genes. Breakage-fusion-bridge cycles and translocations activated oncogenes (BMI1, MIR17HG, TERT, MYC, and MYCN), generating gene amplifications and remodeling regulatory regions. cMCL carried significant higher numbers of structural variants, copy number alterations, and driver changes than nnMCL, with exclusive alterations of ATM in cMCL, whereas TP53 and TERT alterations were slightly enriched in nnMCL. Several drivers had prognostic impact, but only TP53 and MYC aberrations added value independently of genomic complexity. An increasing genomic complexity, together with the presence of breakage-fusion-bridge cycles and high DNA methylation changes related to the proliferative cell history, defines patients with different clinical evolution.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: E.C. is named inventor on 2 patents filed by the National Institutes of Health, National Cancer Institute: “Methods for selecting and treating lymphoma types,” licensed to NanoString Technologies, and “Evaluation of mantle cell lymphoma and methods related thereof.” X.S.P. has ownership interest in DREAMgenics. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Under the cover of mantle cell lymphoma.Blood. 2020 Sep 17;136(12):1378-1379. doi: 10.1182/blood.2020006581. Blood. 2020. PMID: 32941631 No abstract available.
References
-
- In: Swerdlow SH, Campo E, Harris NL, eds., et al.. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues (Revised 4th Edition), Lyon, France: IARC; 2017:
-
- Martin P, Chadburn A, Christos P, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27(8):1209-1213. - PubMed
-
- Martin P, Leonard J. Is there a role for “watch and wait” in patients with mantle cell lymphoma? Semin Hematol. 2011;48(3):189-193. - PubMed
-
- Campo E, Rule S. Mantle cell lymphoma: evolving management strategies. Blood. 2015;125(1):48-55. - PubMed
-
- Fernàndez V, Salamero O, Espinet B, et al. Genomic and gene expression profiling defines indolent forms of mantle cell lymphoma. Cancer Res. 2010;70(4):1408-1418. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
