

Autosomal Dominantly Inherited GREB1L Variants in Individuals with Profound Sensorineural Hearing Impairment
- PMID: 32585897
- PMCID: PMC7349314
- DOI: 10.3390/genes11060687
Autosomal Dominantly Inherited GREB1L Variants in Individuals with Profound Sensorineural Hearing Impairment
Abstract
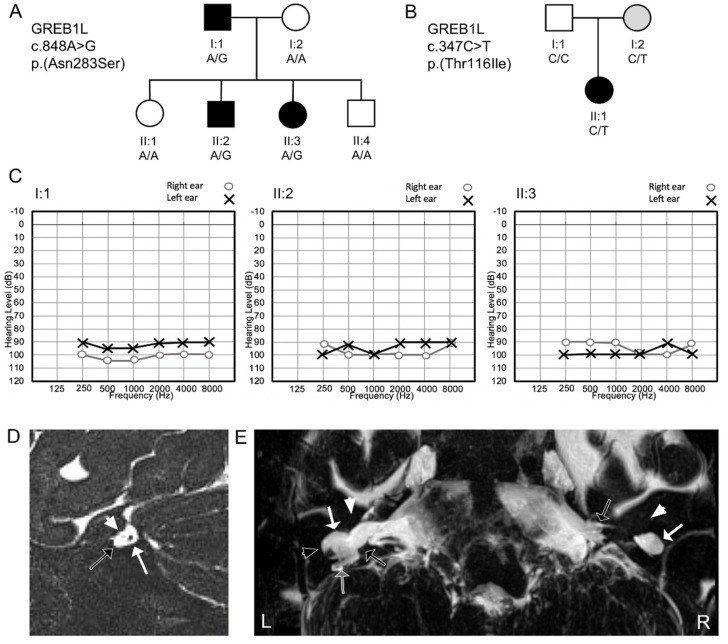
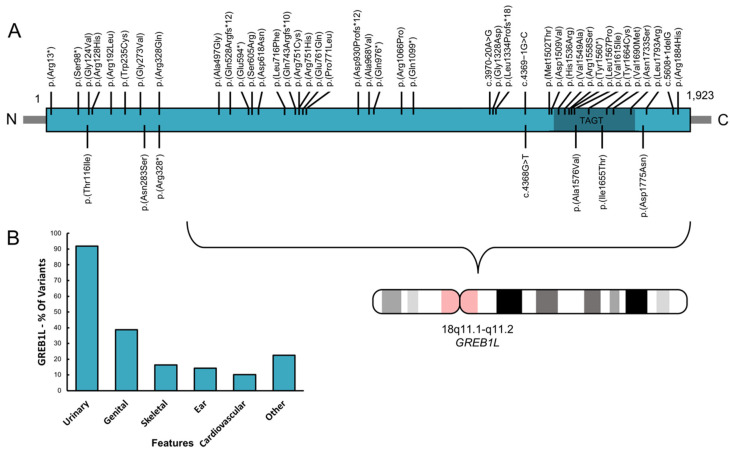
Congenital hearing impairment is a sensory disorder that is genetically highly heterogeneous. By performing exome sequencing in two families with congenital nonsyndromic profound sensorineural hearing loss (SNHL), we identified autosomal dominantly inherited missense variants [p.(Asn283Ser); p.(Thr116Ile)] in GREB1L, a neural crest regulatory molecule. The p.(Thr116Ile) variant was also associated with bilateral cochlear aplasia and cochlear nerve aplasia upon temporal bone imaging, an ultra-rare phenotype previously seen in patients with de novo GREB1L variants. An important role of GREB1L in normal ear development has also been demonstrated by greb1l-/- zebrafish, which show an abnormal sensory epithelia innervation. Last, we performed a review of all disease-associated variation described in GREB1L, as it has also been implicated in renal, bladder and genital malformations. We show that the spectrum of features associated with GREB1L is broad, variable and with a high level of reduced penetrance, which is typically characteristic of neurocristopathies. So far, seven GREB1L variants (14%) have been associated with ear-related abnormalities. In conclusion, these results show that autosomal dominantly inherited variants in GREB1L cause profound SNHL. Furthermore, we provide an overview of the phenotypic spectrum associated with GREB1L variants and strengthen the evidence of the involvement of GREB1L in human hearing.
Keywords: GREB1L; autosomal dominant inheritance; cochlear aplasia; cochlear nerve aplasia; exome sequencing; neural crest; neurocristopathy; profound nonsyndromic hearing impairment.
Conflict of interest statement
The authors have no conflicts of interest related to the work in this manuscript.
Figures
Similar articles
-
De novo variants in GREB1L are associated with non-syndromic inner ear malformations and deafness.Hum Genet. 2018 Jul;137(6-7):459-470. doi: 10.1007/s00439-018-1898-8. Epub 2018 Jun 28. Hum Genet. 2018. PMID: 29955957 Free PMC article.
-
A de novo SIX1 variant in a patient with a rare nonsyndromic cochleovestibular nerve abnormality, cochlear hypoplasia, and bilateral sensorineural hearing loss.Mol Genet Genomic Med. 2019 Dec;7(12):e995. doi: 10.1002/mgg3.995. Epub 2019 Oct 8. Mol Genet Genomic Med. 2019. PMID: 31595699 Free PMC article.
-
Clinical Exome Sequencing Identifies a Novel Mutation of the GREB1L Gene in a Chinese Family with Renal Agenesis.Genet Test Mol Biomarkers. 2020 Aug;24(8):520-526. doi: 10.1089/gtmb.2020.0036. Epub 2020 Jun 25. Genet Test Mol Biomarkers. 2020. PMID: 32598191
-
Recessive LOXHD1 variants cause a prelingual down-sloping hearing loss: genotype-phenotype correlation and three additional children with novel variants.Int J Pediatr Otorhinolaryngol. 2021 Jun;145:110715. doi: 10.1016/j.ijporl.2021.110715. Epub 2021 Apr 20. Int J Pediatr Otorhinolaryngol. 2021. PMID: 33892339 Review.
-
[From gene to disease; non-syndromic, autosomal dominant, low-frequency sensorineural hearing loss (DFNA6/14)].Ned Tijdschr Geneeskd. 2003 Nov 1;147(44):2170-2. Ned Tijdschr Geneeskd. 2003. PMID: 14626834 Review. Dutch.
Cited by
-
A novel variant in DMXL2 gene is associated with autosomal dominant non-syndromic hearing impairment (DFNA71) in a Cameroonian family.Exp Biol Med (Maywood). 2021 Jul;246(13):1524-1532. doi: 10.1177/1535370221999746. Epub 2021 Mar 9. Exp Biol Med (Maywood). 2021. PMID: 33715530 Free PMC article.
-
Major Contribution of GREB1L Alterations to Severe Inner Ear Malformation Largely in a Non-mendelian Fashion.Clin Exp Otorhinolaryngol. 2022 Feb;15(1):115-118. doi: 10.21053/ceo.2021.01935. Epub 2022 Jan 12. Clin Exp Otorhinolaryngol. 2022. PMID: 35012281 Free PMC article. No abstract available.
-
Bi-Allelic Novel Variants in CLIC5 Identified in a Cameroonian Multiplex Family with Non-Syndromic Hearing Impairment.Genes (Basel). 2020 Oct 23;11(11):1249. doi: 10.3390/genes11111249. Genes (Basel). 2020. PMID: 33114113 Free PMC article.
-
A novel autosomal dominant GREB1L variant associated with non-syndromic hearing impairment in Ghana.BMC Med Genomics. 2022 Nov 10;15(1):237. doi: 10.1186/s12920-022-01391-w. BMC Med Genomics. 2022. PMID: 36357908 Free PMC article.
-
Genome Sequencing and Transcriptome Profiling in Twins Discordant for Mayer-Rokitansky-Küster-Hauser Syndrome.J Clin Med. 2022 Sep 23;11(19):5598. doi: 10.3390/jcm11195598. J Clin Med. 2022. PMID: 36233463 Free PMC article.
References
-
- Shearer A.E., Hildebrand M.S., Smith R.J. Hereditary hearing loss and deafness overview. In: Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J., Stephens K., Amemiya A., editors. GeneReviews®. University of Washington; Seattle, WA, USA: 1993.
-
- Chakchouk I., Zhang D., Zhang Z., Francioli L.C., Santos-Cortez R.L.P., Schrauwen I., Leal S.M. Disparities in discovery of pathogenic variants for autosomal recessive non-syndromic hearing impairment by ancestry. Eur. J. Hum. Genet. 2019;27:1456–1465. doi: 10.1038/s41431-019-0417-2. - DOI - PMC - PubMed
-
- Kaplan A.B., Kozin E.D., Puram S.V., Owoc M.S., Shah P.V., Hight A.E., Sethi R.K.V., Remenschneider A.K., Lee D.J. Auditory brainstem implant candidacy in the United States in children 0–17 years old. Int. J. Pediatr. Otorhinolaryngol. 2015;79:310–315. doi: 10.1016/j.ijporl.2014.11.023. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
