

The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications
- PMID: 32586214
- PMCID: PMC7386875
- DOI: 10.1161/CIRCRESAHA.120.317447
The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications
Abstract
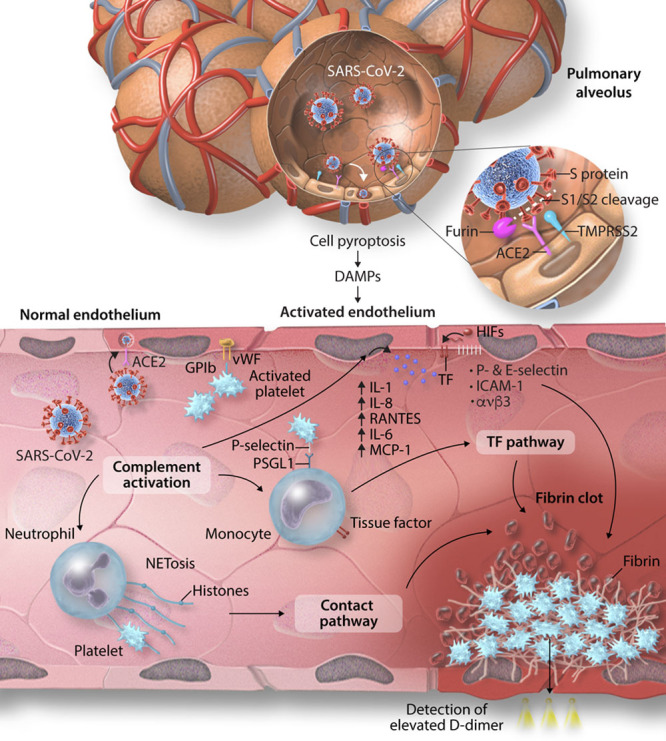
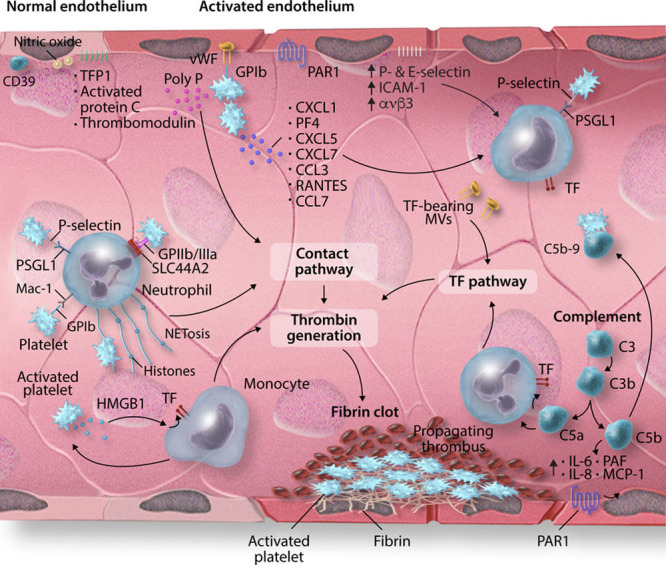
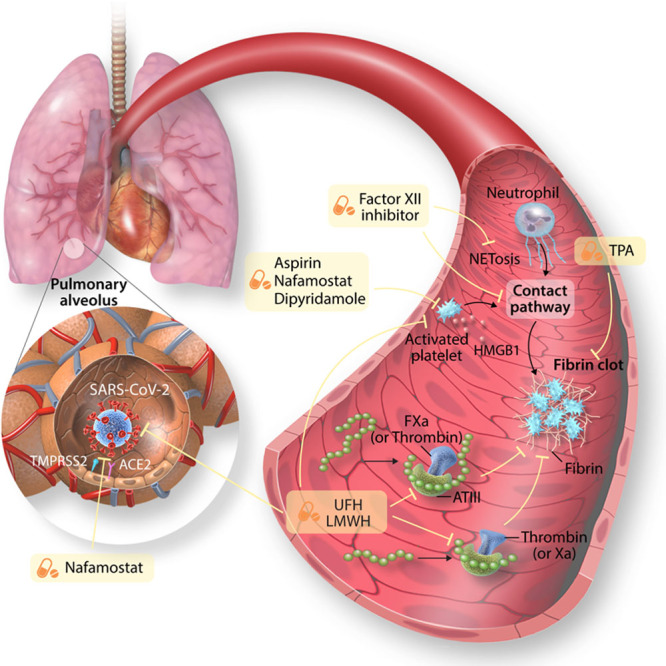
The recent emergence of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the ensuing global pandemic has presented a health emergency of unprecedented magnitude. Recent clinical data has highlighted that coronavirus disease 2019 (COVID-19) is associated with a significant risk of thrombotic complications ranging from microvascular thrombosis, venous thromboembolic disease, and stroke. Importantly, thrombotic complications are markers of severe COVID-19 and are associated with multiorgan failure and increased mortality. The evidence to date supports the concept that the thrombotic manifestations of severe COVID-19 are due to the ability of SARS-CoV-2 to invade endothelial cells via ACE-2 (angiotensin-converting enzyme 2), which is expressed on the endothelial cell surface. However, in patients with COVID-19 the subsequent endothelial inflammation, complement activation, thrombin generation, platelet, and leukocyte recruitment, and the initiation of innate and adaptive immune responses culminate in immunothrombosis, ultimately causing (micro)thrombotic complications, such as deep vein thrombosis, pulmonary embolism, and stroke. Accordingly, the activation of coagulation (eg, as measured with plasma D-dimer) and thrombocytopenia have emerged as prognostic markers in COVID-19. Given thrombotic complications are central determinants of the high mortality rate in COVID-19, strategies to prevent thrombosis are of critical importance. Several antithrombotic drugs have been proposed as potential therapies to prevent COVID-19-associated thrombosis, including heparin, FXII inhibitors, fibrinolytic drugs, nafamostat, and dipyridamole, many of which also possess pleiotropic anti-inflammatory or antiviral effects. The growing awareness and mechanistic understanding of the prothrombotic state of COVID-19 patients are driving efforts to more stringent diagnostic screening for thrombotic complications and to the early institution of antithrombotic drugs, for both the prevention and therapy of thrombotic complications. The shifting paradigm of diagnostic and treatment strategies holds significant promise to reduce the burden of thrombotic complications and ultimately improve the prognosis for patients with COVID-19.
Keywords: coronavirus; mortality; stroke; thrombosis; viruses.
Figures

References
-
- Lam TT, Jia N, Zhang YW, Shum MH, Jiang JF, Zhu HC, Tong YG, Shi YX, Ni XB, Liao YS, et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature. 2020 doi: 10.1038/s41586-020-2169-0. - PubMed
-
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020181271–280.e8doi: 10.1016/j.cell.2020.02.052 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
