

Exploring whole-genome duplicate gene retention with complex genetic interaction analysis
- PMID: 32586993
- PMCID: PMC7539174
- DOI: 10.1126/science.aaz5667
Exploring whole-genome duplicate gene retention with complex genetic interaction analysis
Abstract
Whole-genome duplication has played a central role in the genome evolution of many organisms, including the human genome. Most duplicated genes are eliminated, and factors that influence the retention of persisting duplicates remain poorly understood. We describe a systematic complex genetic interaction analysis with yeast paralogs derived from the whole-genome duplication event. Mapping of digenic interactions for a deletion mutant of each paralog, and of trigenic interactions for the double mutant, provides insight into their roles and a quantitative measure of their functional redundancy. Trigenic interaction analysis distinguishes two classes of paralogs: a more functionally divergent subset and another that retained more functional overlap. Gene feature analysis and modeling suggest that evolutionary trajectories of duplicated genes are dictated by combined functional and structural entanglement factors.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
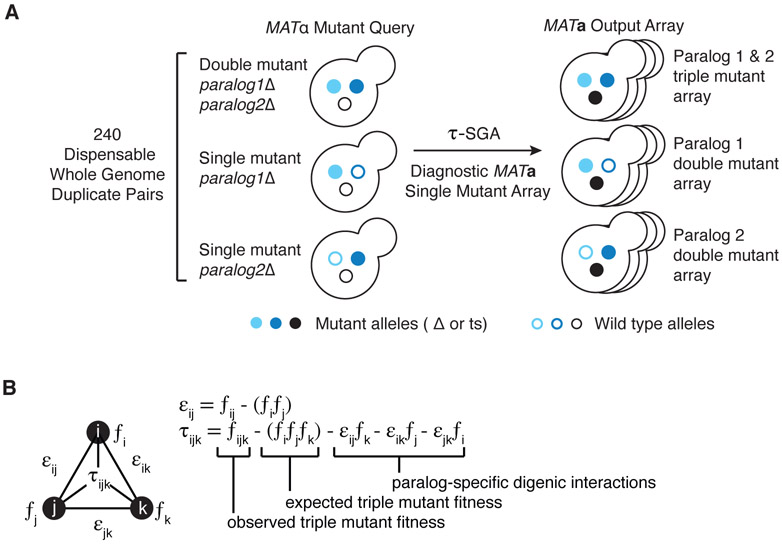
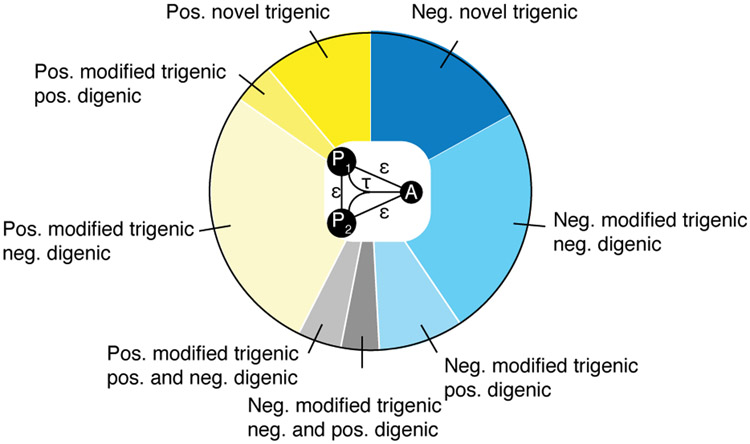
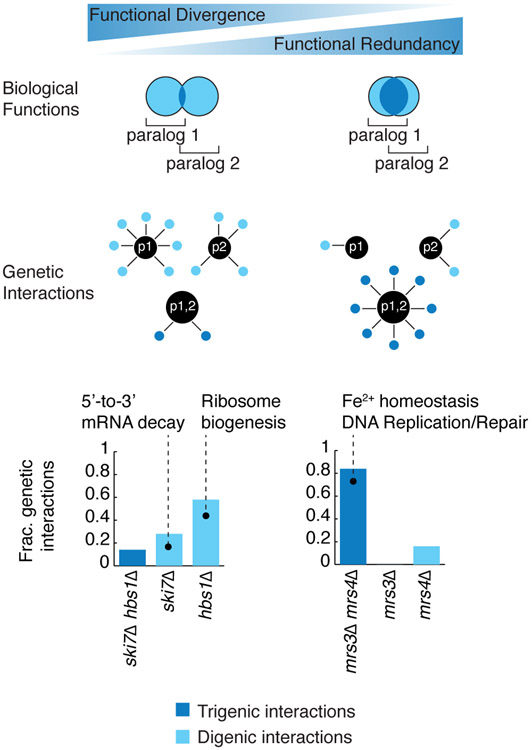
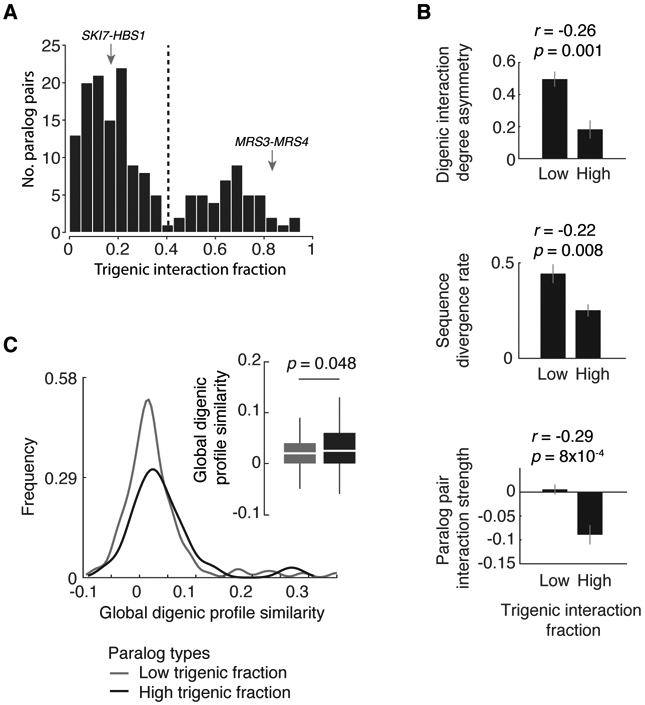
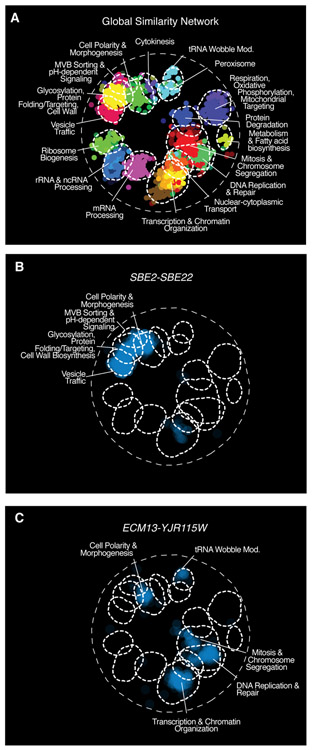
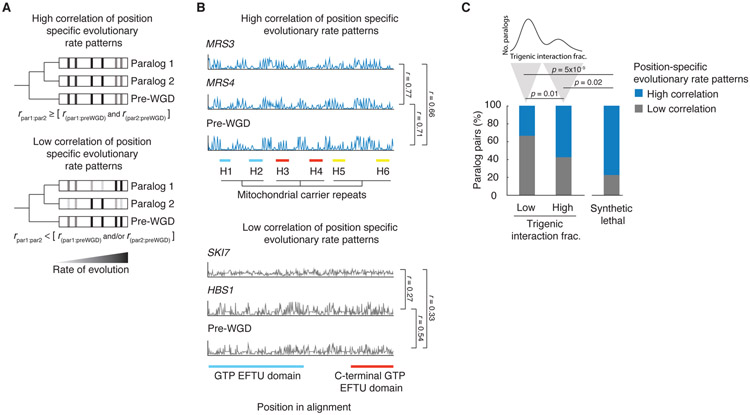
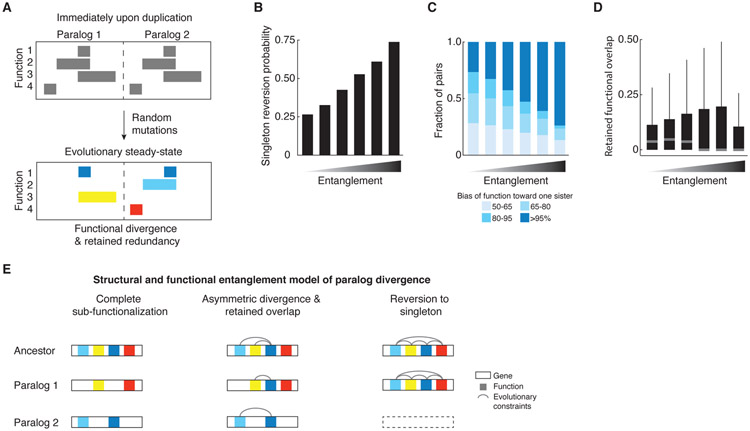
Comment in
-
Evolution after genome duplication.Science. 2020 Jun 26;368(6498):1424-1425. doi: 10.1126/science.abc1796. Science. 2020. PMID: 32587005 No abstract available.
References
-
- Bowers JE, Chapman BA, Rong J, Paterson AH, Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422, 433–438 (2003). - PubMed
-
- Eichler EE, Recent duplication, domain accretion and the dynamic mutation of the human genome. Trends Genet 17, 661–669 (2001). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
