

Early infantile epileptic encephalopathy due to biallelic pathogenic variants in PIGQ: Report of seven new subjects and review of the literature
- PMID: 32588908
- PMCID: PMC7689772
- DOI: 10.1002/jimd.12278
Early infantile epileptic encephalopathy due to biallelic pathogenic variants in PIGQ: Report of seven new subjects and review of the literature
Erratum in
-
Erratum.J Inherit Metab Dis. 2023 Jan;46(1):156. doi: 10.1002/jimd.12362. J Inherit Metab Dis. 2023. PMID: 36646502 Free PMC article. No abstract available.
Abstract
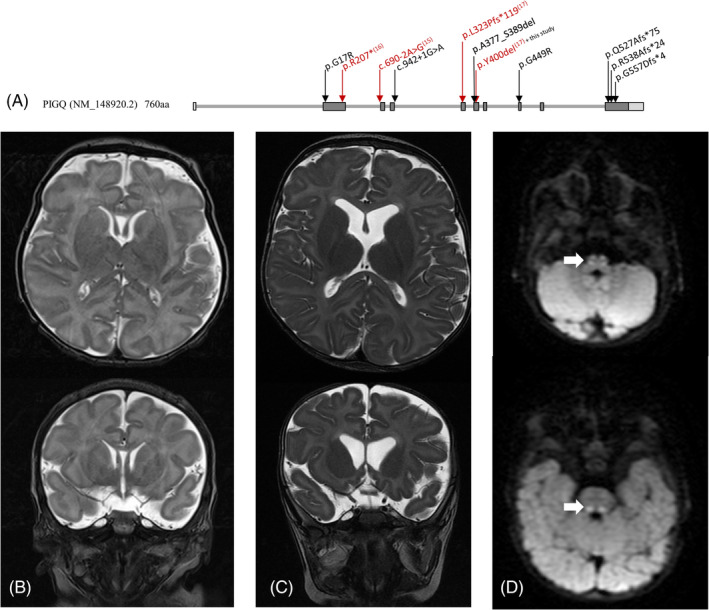
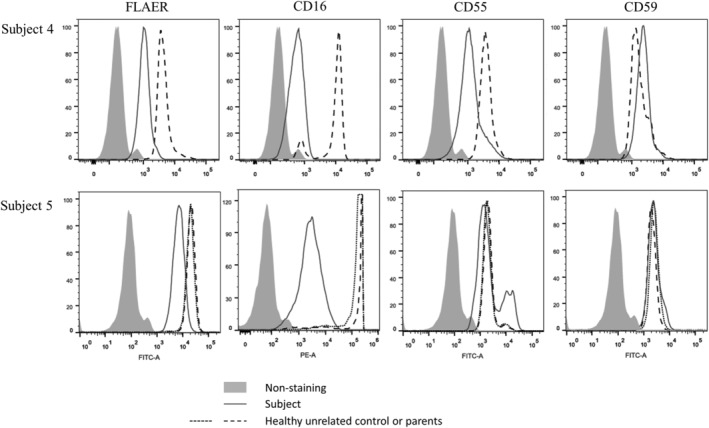
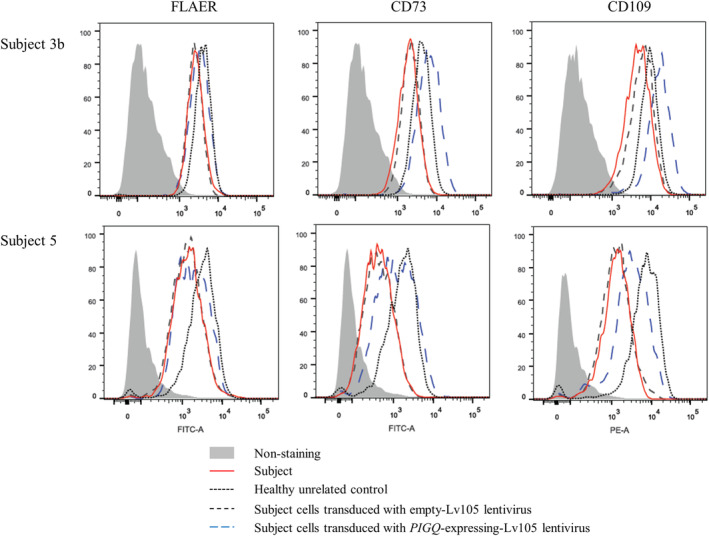
We investigated seven children from six families to expand the phenotypic spectrum associated with an early infantile epileptic encephalopathy caused by biallelic pathogenic variants in the phosphatidylinositol glycan anchor biosynthesis class Q (PIGQ) gene. The affected children were all identified by clinical or research exome sequencing. Clinical data, including EEGs and MRIs, was comprehensively reviewed and flow cytometry and transfection experiments were performed to investigate PIGQ function. Pathogenic biallelic PIGQ variants were associated with increased mortality. Epileptic seizures, axial hypotonia, developmental delay and multiple congenital anomalies were consistently observed. Seizure onset occurred between 2.5 months and 7 months of age and varied from treatable seizures to recurrent episodes of status epilepticus. Gastrointestinal issues were common and severe, two affected individuals had midgut volvulus requiring surgical correction. Cardiac anomalies including arrythmias were observed. Flow cytometry using granulocytes and fibroblasts from affected individuals showed reduced expression of glycosylphosphatidylinositol (GPI)-anchored proteins. Transfection of wildtype PIGQ cDNA into patient fibroblasts rescued this phenotype. We expand the phenotypic spectrum of PIGQ-related disease and provide the first functional evidence in human cells of defective GPI-anchoring due to pathogenic variants in PIGQ.
Keywords: GPI; IGD; PIGQ; epileptic encephalopathy; exome sequencing; rare diseases.
© 2020 The Authors. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Devon L. Johnstone, Thi Tuyet Mai Nguyen, Jessica Zambonin, Kristin Kernohan, Anik St‐Denis, Nissan V. Baratang, Taila Hartley, Michael T. Geraghty, Julie Richer, Jacek Majewski, Eric Bareke, Andrea Guerin, Manuela Pendziwiat, Loren D.M. Pena, Hilde M. H. Braakman, Karen W. Gripp, Andrew C. Edmondson, Miao He, Rebecca C. Spillmann, Erik A. Eklund, Allan Bayat, Hugh J.McMillan, Kym M. Boycott, and Philippe M. Campeau declare that they have no conflict of interest.
Figures
References
-
- Bellai‐Dussault K, Nguyen TTM, Baratang NV, Jimenez‐Cruz DA, Campeau PM. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin Genet. 2019;95(1):112‐121. - PubMed
-
- Nozaki M, Ohishi K, Yamada N, Kinoshita T, Nagy A, Takeda J. Developmental abnormalities of glycosylphosphatidylinositol‐anchor‐deficient embryos revealed by Cre/loxP system. Lab Invest. 1999;79(3):293‐299. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
