

The Renin-Angiotensin-Aldosterone System (RAAS) Is One of the Effectors by Which Vascular Endothelial Growth Factor (VEGF)/Anti-VEGF Controls the Endothelial Cell Barrier
- PMID: 32590003
- PMCID: PMC7538812
- DOI: 10.1016/j.ajpath.2020.06.004
The Renin-Angiotensin-Aldosterone System (RAAS) Is One of the Effectors by Which Vascular Endothelial Growth Factor (VEGF)/Anti-VEGF Controls the Endothelial Cell Barrier
Abstract
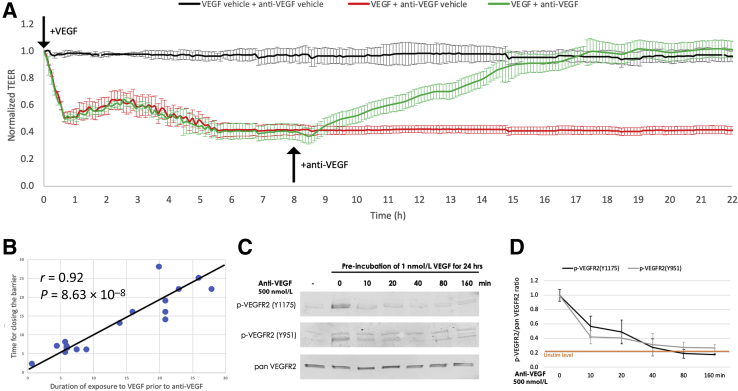
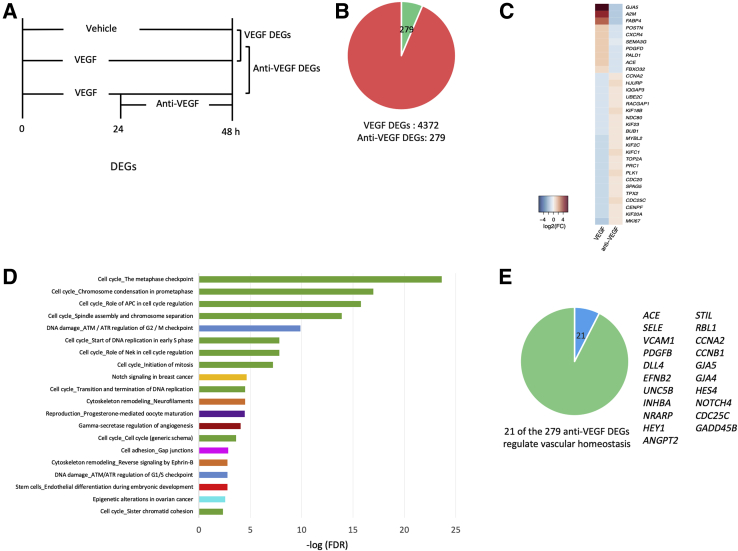
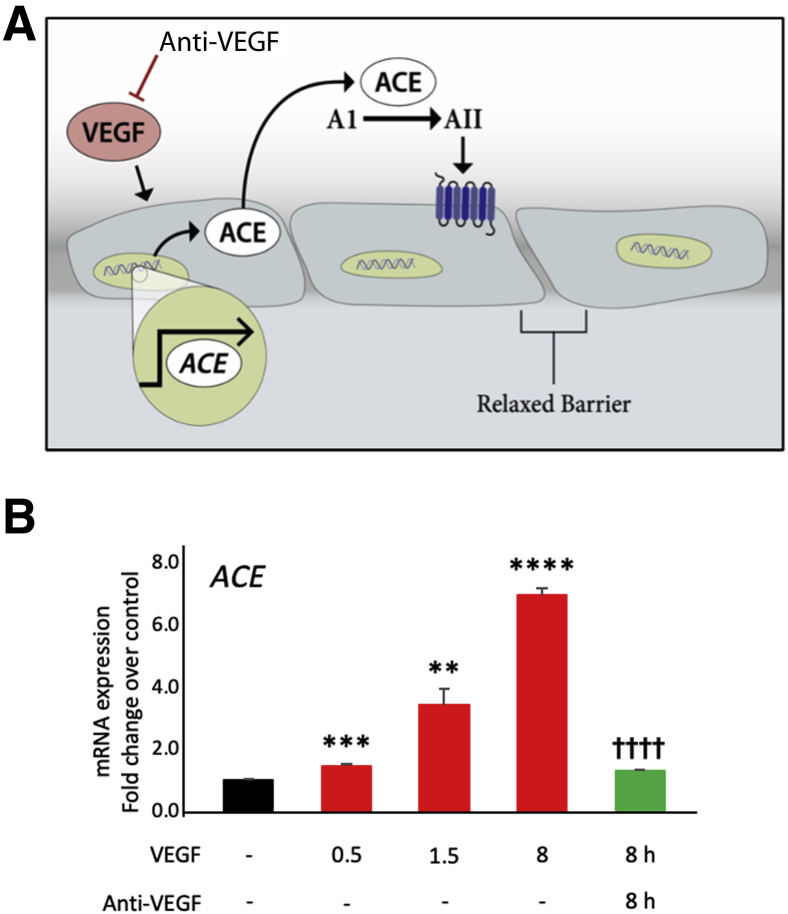
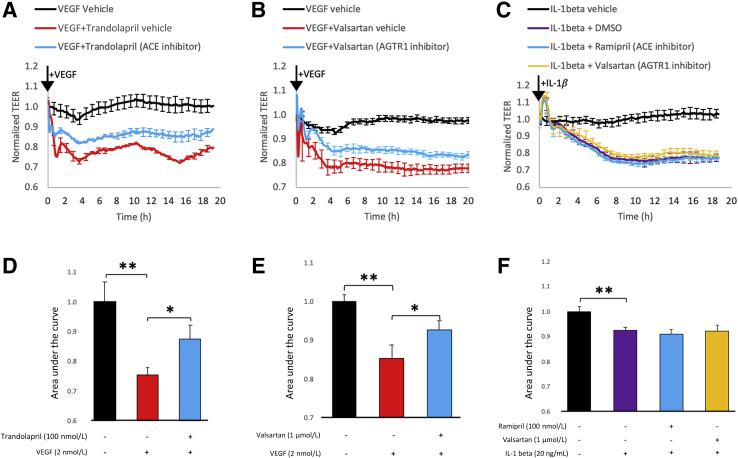
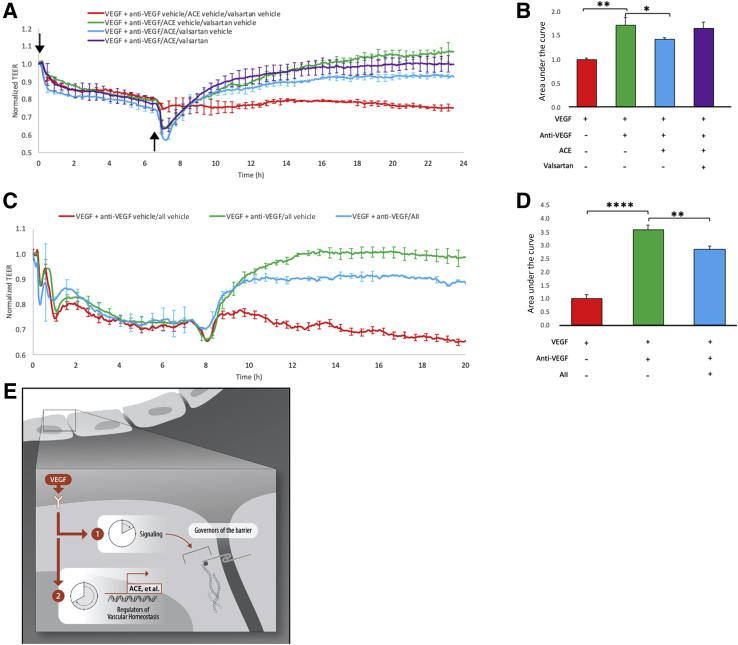
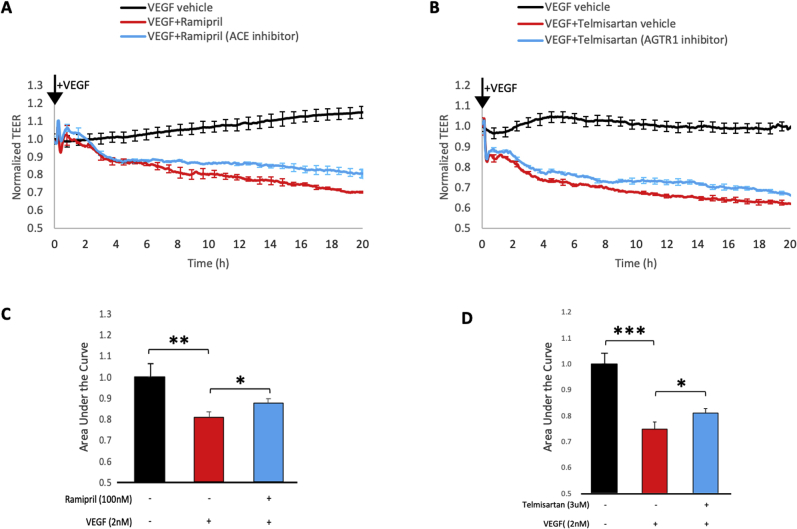
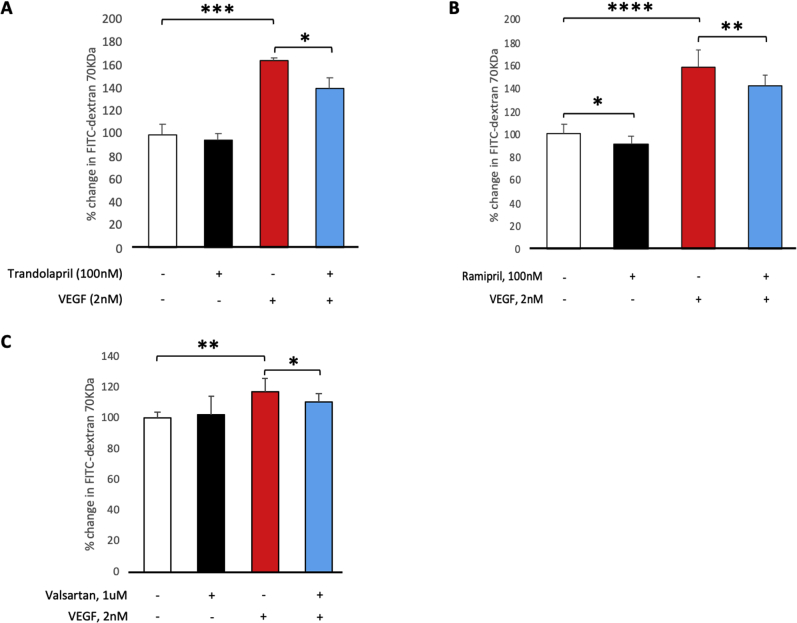
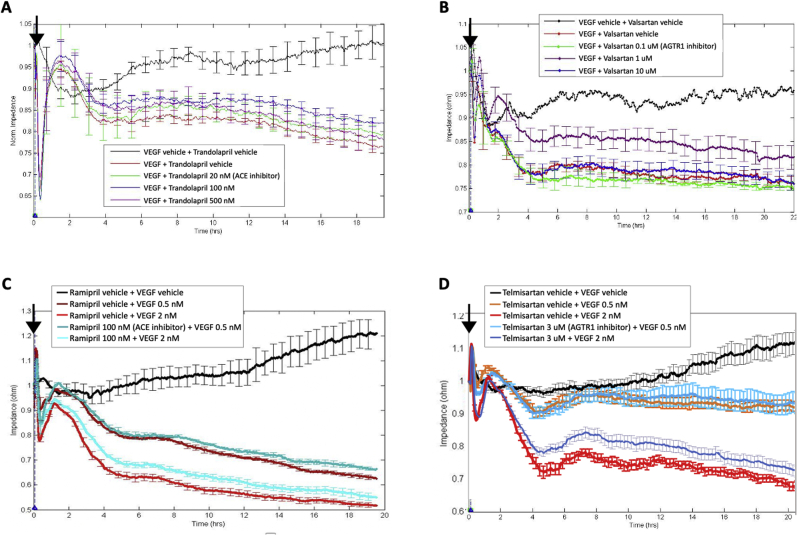
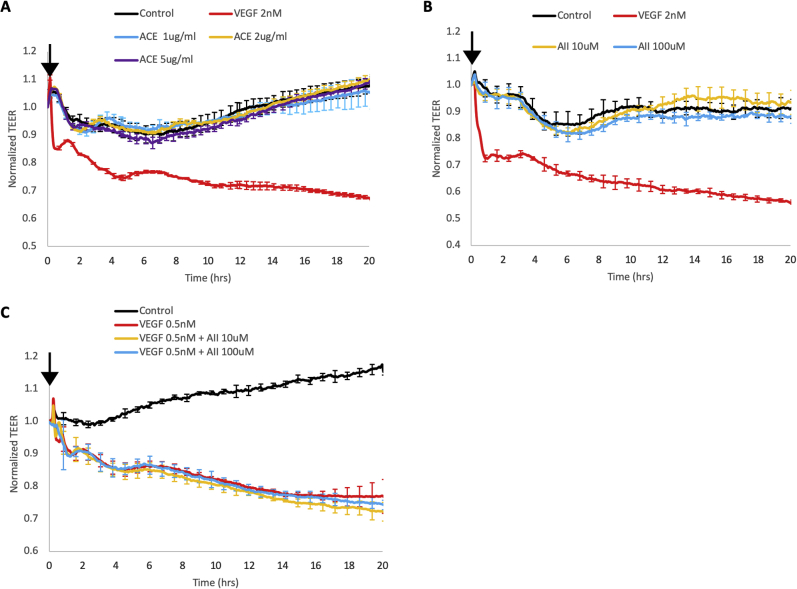
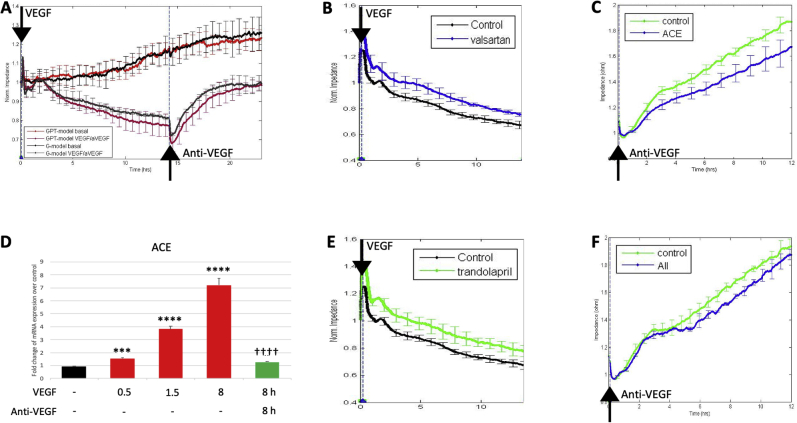
Leakage of retinal blood vessels, which is an essential element of diabetic retinopathy, is driven by chronic elevation of vascular endothelial growth factor (VEGF). VEGF quickly relaxes the endothelial cell barrier by triggering signaling events that post-translationally modify pre-existing components of intercellular junctions. VEGF also changes expression of genes that are known to regulate barrier function. Our goal was to identify effectors by which VEGF and anti-VEGF control the endothelial cell barrier in cells that were chronically exposed to VEGF (hours instead of minutes). The duration of VEGF exposure influenced both barrier relaxation and anti-VEGF-mediated closure. Most VEGF-induced changes in gene expression were not reversed by anti-VEGF. Those that were constitute VEGF effectors that are targets of anti-VEGF. Pursuit of such candidates revealed that VEGF used multiple, nonredundant effectors to relax the barrier in cells that were chronically exposed to VEGF. One such effector was angiotensin-converting enzyme, which is a member of the renin-angiotensin-aldosterone system (RAAS). Pharmacologically antagonizing either the angiotensin-converting enzyme or the receptor for angiotensin II attenuated VEGF-mediated relaxation of the barrier. Finally, activating the RAAS reduced the efficacy of anti-VEGF. These discoveries provide a plausible mechanistic explanation for the long-standing appreciation that RAAS inhibitors are beneficial for patients with diabetic retinopathy and suggest that antagonizing the RAAS improves patients' responsiveness to anti-VEGF.
Copyright © 2020 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Blockade of angiotensin II attenuates VEGF-mediated blood-retinal barrier breakdown in diabetic retinopathy.J Cereb Blood Flow Metab. 2009 Mar;29(3):621-8. doi: 10.1038/jcbfm.2008.154. Epub 2008 Dec 24. J Cereb Blood Flow Metab. 2009. PMID: 19107135
-
Effects of angiotensin-converting enzyme inhibitors and beta-adrenergic blockers on retinal vascular endothelial growth factor expression in rat diabetic retinopathy.Exp Eye Res. 2007 Apr;84(4):745-52. doi: 10.1016/j.exer.2006.12.013. Epub 2006 Dec 31. Exp Eye Res. 2007. PMID: 17303121
-
Erythropoietin maintains VE-cadherin expression and barrier function in experimental diabetic retinopathy via inhibiting VEGF/VEGFR2/Src signaling pathway.Life Sci. 2020 Oct 15;259:118273. doi: 10.1016/j.lfs.2020.118273. Epub 2020 Aug 12. Life Sci. 2020. PMID: 32800831
-
Angiotensin II and aldosterone in retinal vasculopathy and inflammation.Exp Eye Res. 2019 Oct;187:107766. doi: 10.1016/j.exer.2019.107766. Epub 2019 Aug 16. Exp Eye Res. 2019. PMID: 31425690 Review.
-
The renin-angiotensin system and diabetic retinopathy.Klin Wochenschr. 1992;69 Suppl 29:25-7. Klin Wochenschr. 1992. PMID: 1614187 Review.
Cited by
-
Effects of regular, glulisine, and aspart insulin on vascular endothelial growth factor and angiotensinogen expression in hyperglycemic retinal pigment epithelial (RPE) and human retinal endothelial cells (HRECs).Front Ophthalmol (Lausanne). 2025 May 29;5:1570232. doi: 10.3389/fopht.2025.1570232. eCollection 2025. Front Ophthalmol (Lausanne). 2025. PMID: 40510890 Free PMC article.
-
Metabolic Dysregulation in Idiopathic Pulmonary Fibrosis.Int J Mol Sci. 2020 Aug 7;21(16):5663. doi: 10.3390/ijms21165663. Int J Mol Sci. 2020. PMID: 32784632 Free PMC article. Review.
-
Different responses of the MIO‑M1 Mueller cell line to angiotensin II under hyperglycemic or hypoxic conditions.Biomed Rep. 2023 Aug 7;19(3):62. doi: 10.3892/br.2023.1644. eCollection 2023 Sep. Biomed Rep. 2023. PMID: 37614982 Free PMC article.
-
Genetics of Diabetic Retinopathy, a Leading Cause of Irreversible Blindness in the Industrialized World.Genes (Basel). 2021 Jul 31;12(8):1200. doi: 10.3390/genes12081200. Genes (Basel). 2021. PMID: 34440374 Free PMC article. Review.
-
Modified Qing' e Pills exerts anti-osteoporosis effects and prevents bone loss by enhancing type H blood vessel formation.Front Endocrinol (Lausanne). 2022 Sep 6;13:998971. doi: 10.3389/fendo.2022.998971. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36147560 Free PMC article.
References
-
- Stitt A.W., Curtis T.M., Chen M., Medina R.J., McKay G.J., Jenkins A., Gardiner T.A., Lyons T.J., Hammes H.P., Simo R., Lois N. The progress in understanding and treatment of diabetic retinopathy. Prog Retin Eye Res. 2016;51:156–186. - PubMed
-
- Cheung N., Mitchell P., Wong T.Y. Diabetic retinopathy. Lancet. 2010;376:124–136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
