

KIAA1109 gene mutation in surviving patients with Alkuraya-Kučinskas syndrome: a review of literature
- PMID: 32590954
- PMCID: PMC7318400
- DOI: 10.1186/s12881-020-01074-2
KIAA1109 gene mutation in surviving patients with Alkuraya-Kučinskas syndrome: a review of literature
Abstract
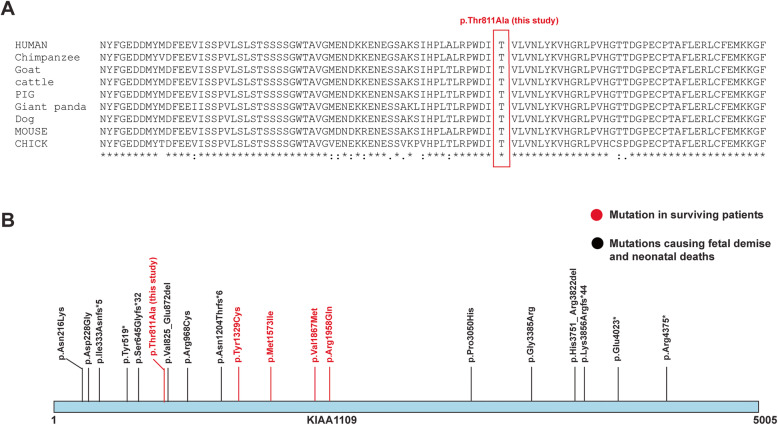
Background: Alkuraya-Kučinskas syndrome is an autosomal recessive disorder characterized by brain abnormalities associated with cerebral parenchymal underdevelopment, arthrogryposis, club foot and global developmental delay. KIAA1109, a functionally uncharacterized gene is identified as the molecular cause for Alkuraya-Kučinskas syndrome. Most of the reported mutations in KIAA1109 gene result in premature termination of pregnancies or neonatal deaths while a few mutations have been reported in surviving patients with global developmental delay and intellectual disability. To our knowledge, only three surviving patients from two families have been reported with missense variants in KIAA1109. In this study, we describe four surviving patients from two related families (a multiplex family) with global developmental delay and mild to severe intellectual disability with no other systemic manifestations. There were no miscarriages or neonatal deaths reported in these families.
Methods: X-chromosome exome panel sequencing was carried out in one patient and whole exome sequencing was carried out on the remaining three affected individuals and the unaffected father of the index family. Data analysis was carried out followed by variant filtering and segregation analysis. Sanger sequencing was carried out to validate the segregation of mutation in all four affected siblings and unaffected parents from both families.
Results: A novel homozygous missense mutation in a conserved region of KIAA1109 protein was identified. Sanger sequencing confirmed the segregation of mutation in both families in an autosomal recessive fashion.
Conclusion: Our study is the second study reporting a KIAA1109 variant in surviving patients with Alkuraya-Kučinskas syndrome. Our study expands the spectrum of phenotypic features and mutations associated with Alkuraya-Kučinskas syndrome.
Keywords: Arthrogryposis; Club foot; Developmental delay; KIAA clones; Mental retardation; Miscarriages; Neonatal death; Premature termination of pregnancy; Prenatal diagnosis.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Gueneau L, Fish RJ, Shamseldin HE, Voisin N, Tran Mau-Them F, Preiksaitiene E, Monroe GR, Lai A, Putoux A, Allias F, et al. KIAA1109 variants are associated with a severe disorder of brain development and Arthrogryposis. Am J Hum Genet. 2018;102(1):116–132. doi: 10.1016/j.ajhg.2017.12.002. - DOI - PMC - PubMed
-
- Shrine N, Portelli MA, John C, Soler Artigas M, Bennett N, Hall R, Lewis J, Henry AP, Billington CK, Ahmad A, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med. 2019;7(1):20–34. doi: 10.1016/S2213-2600(18)30389-8. - DOI - PMC - PubMed
-
- Glas J, Stallhofer J, Ripke S, Wetzke M, Pfennig S, Klein W, Epplen JT, Griga T, Schiemann U, Lacher M, et al. Novel genetic risk markers for ulcerative colitis in the IL2/IL21 region are in epistasis with IL23R and suggest a common genetic background for ulcerative colitis and celiac disease. Am J Gastroenterol. 2009;104(7):1737–1744. doi: 10.1038/ajg.2009.163. - DOI - PubMed
-
- Plaza-Izurieta L, Castellanos-Rubio A, Irastorza I, Fernandez-Jimenez N, Gutierrez G, Cegec BJR. Revisiting genome wide association studies (GWAS) in coeliac disease: replication study in Spanish population and expression analysis of candidate genes. J Med Genet. 2011;48(7):493–496. doi: 10.1136/jmg.2011.089714. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
