

Detection of copy-number variations from NGS data using read depth information: a diagnostic performance evaluation
- PMID: 32591635
- PMCID: PMC7852510
- DOI: 10.1038/s41431-020-0672-2
Detection of copy-number variations from NGS data using read depth information: a diagnostic performance evaluation
Abstract
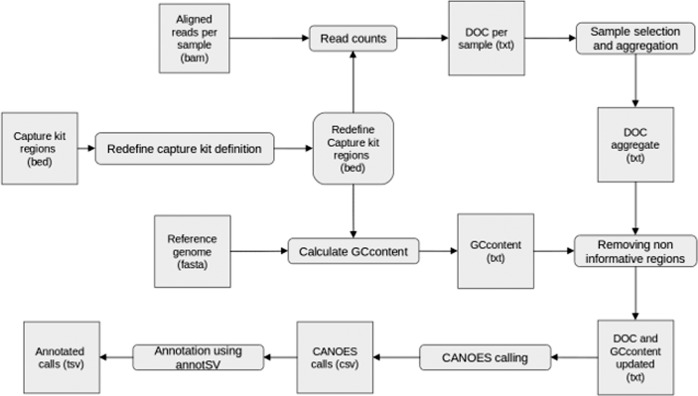
The detection of copy-number variations (CNVs) from NGS data is underexploited as chip-based or targeted techniques are still commonly used. We assessed the performances of a workflow centered on CANOES, a bioinformatics tool based on read depth information. We applied our workflow to gene panel (GP) and whole-exome sequencing (WES) data, and compared CNV calls to quantitative multiplex PCR of short fluorescent fragments (QMSPF) or array comparative genomic hybridization (aCGH) results. From GP data of 3776 samples, we reached an overall positive predictive value (PPV) of 87.8%. This dataset included a complete comprehensive QMPSF comparison of four genes (60 exons) on which we obtained 100% sensitivity and specificity. From WES data, we first compared 137 samples with aCGH and filtered comparable events (exonic CNVs encompassing enough aCGH probes) and obtained an 87.25% sensitivity. The overall PPV was 86.4% following the targeted confirmation of candidate CNVs from 1056 additional WES. In addition, our CANOES-centered workflow on WES data allowed the detection of CNVs with a resolution of single exons, allowing the detection of CNVs that were missed by aCGH. Overall, switching to an NGS-only approach should be cost-effective as it allows a reduction in overall costs together with likely stable diagnostic yields. Our bioinformatics pipeline is available at: https://gitlab.bioinfo-diag.fr/nc4gpm/canoes-centered-workflow .
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
