

BnpC: Bayesian non-parametric clustering of single-cell mutation profiles
- PMID: 32592465
- PMCID: PMC7750970
- DOI: 10.1093/bioinformatics/btaa599
BnpC: Bayesian non-parametric clustering of single-cell mutation profiles
Abstract
Motivation: The high resolution of single-cell DNA sequencing (scDNA-seq) offers great potential to resolve intratumor heterogeneity (ITH) by distinguishing clonal populations based on their mutation profiles. However, the increasing size of scDNA-seq datasets and technical limitations, such as high error rates and a large proportion of missing values, complicate this task and limit the applicability of existing methods.
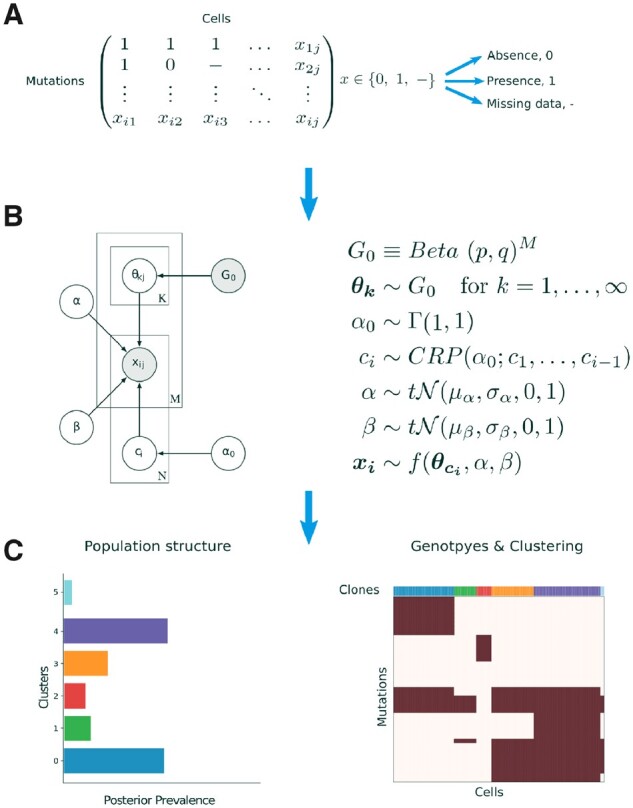
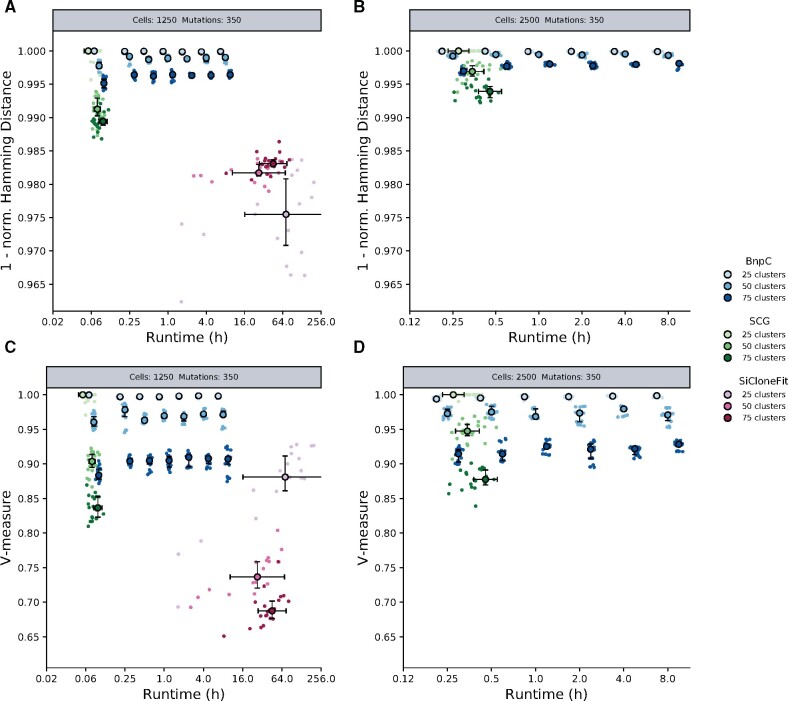
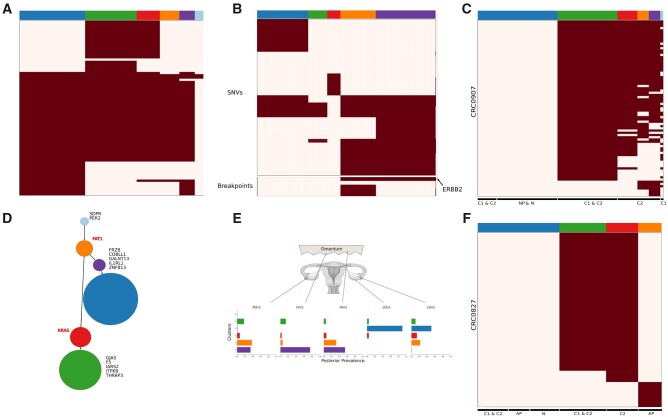
Results: Here, we introduce BnpC, a novel non-parametric method to cluster individual cells into clones and infer their genotypes based on their noisy mutation profiles. We benchmarked our method comprehensively against state-of-the-art methods on simulated data using various data sizes, and applied it to three cancer scDNA-seq datasets. On simulated data, BnpC compared favorably against current methods in terms of accuracy, runtime and scalability. Its inferred genotypes were the most accurate, especially on highly heterogeneous data, and it was the only method able to run and produce results on datasets with 5000 cells. On tumor scDNA-seq data, BnpC was able to identify clonal populations missed by the original cluster analysis but supported by Supplementary Experimental Data. With ever growing scDNA-seq datasets, scalable and accurate methods such as BnpC will become increasingly relevant, not only to resolve ITH but also as a preprocessing step to reduce data size.
Availability and implementation: BnpC is freely available under MIT license at https://github.com/cbg-ethz/BnpC.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
-
- Burrell R.A. et al. (2013) The causes and consequences of genetic heterogeneity in cancer evolution. Nature, 501, 338–345. - PubMed
-
- Ciccolella S. et al. (2019) Benchmarking clustering methods for single cell sequencing cancer data. bioRxiv.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
