

Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer
- PMID: 32593127
- PMCID: PMC7322178
- DOI: 10.1016/j.redox.2020.101510
Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer
Abstract
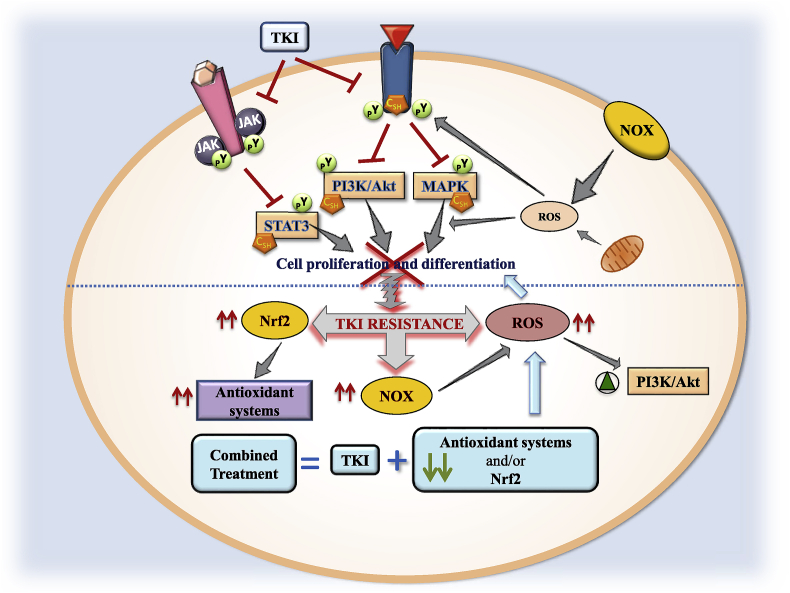
Cancer cells have unlimited replicative potential, insensitivity to growth-inhibitory signals, evasion of apoptosis, cellular stress, and sustained angiogenesis, invasiveness and metastatic potential. Cancer cells adequately adapt cell metabolism and integrate several intracellular and redox signaling to promote cell survival in an inflammatory and hypoxic microenvironment in order to maintain/expand tumor phenotype. The administration of tyrosine kinase inhibitor (TKI) constitutes the recommended therapeutic strategy in different malignancies at advanced stages. There are important interrelationships between cell stress, redox status, mitochondrial function, metabolism and cellular signaling pathways leading to cell survival/death. The induction of apoptosis and cell cycle arrest widely related to the antitumoral properties of TKIs result from tightly controlled events involving different cellular compartments and signaling pathways. The aim of the present review is to update the most relevant studies dealing with the impact of TKI treatment on cell function. The induction of endoplasmic reticulum (ER) stress and Ca2+ disturbances, leading to alteration of mitochondrial function, redox status and phosphatidylinositol 3-kinase (PI3K)-protein kinase B (Akt)-mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) signaling pathways that involve cell metabolism reprogramming in cancer cells will be covered. Emphasis will be given to studies that identify key components of the integrated molecular pattern including receptor tyrosine kinase (RTK) downstream signaling, cell death and mitochondria-related events that appear to be involved in the resistance of cancer cells to TKI treatments.
Keywords: Autophagy; Cell death; Endoplasmic reticulum stress; PGC-1α; Redox status; mTOR.
Copyright © 2020 The Authors. Published by Elsevier B.V. All rights reserved.
Figures





References
-
- Lomax M.E., Folkes L.K., O'Neill P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin. Oncol. 2013;25:578–585. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
