

Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy
- PMID: 32597196
- PMCID: PMC7484440
- DOI: 10.1161/CIRCULATIONAHA.119.043319
Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy
Abstract
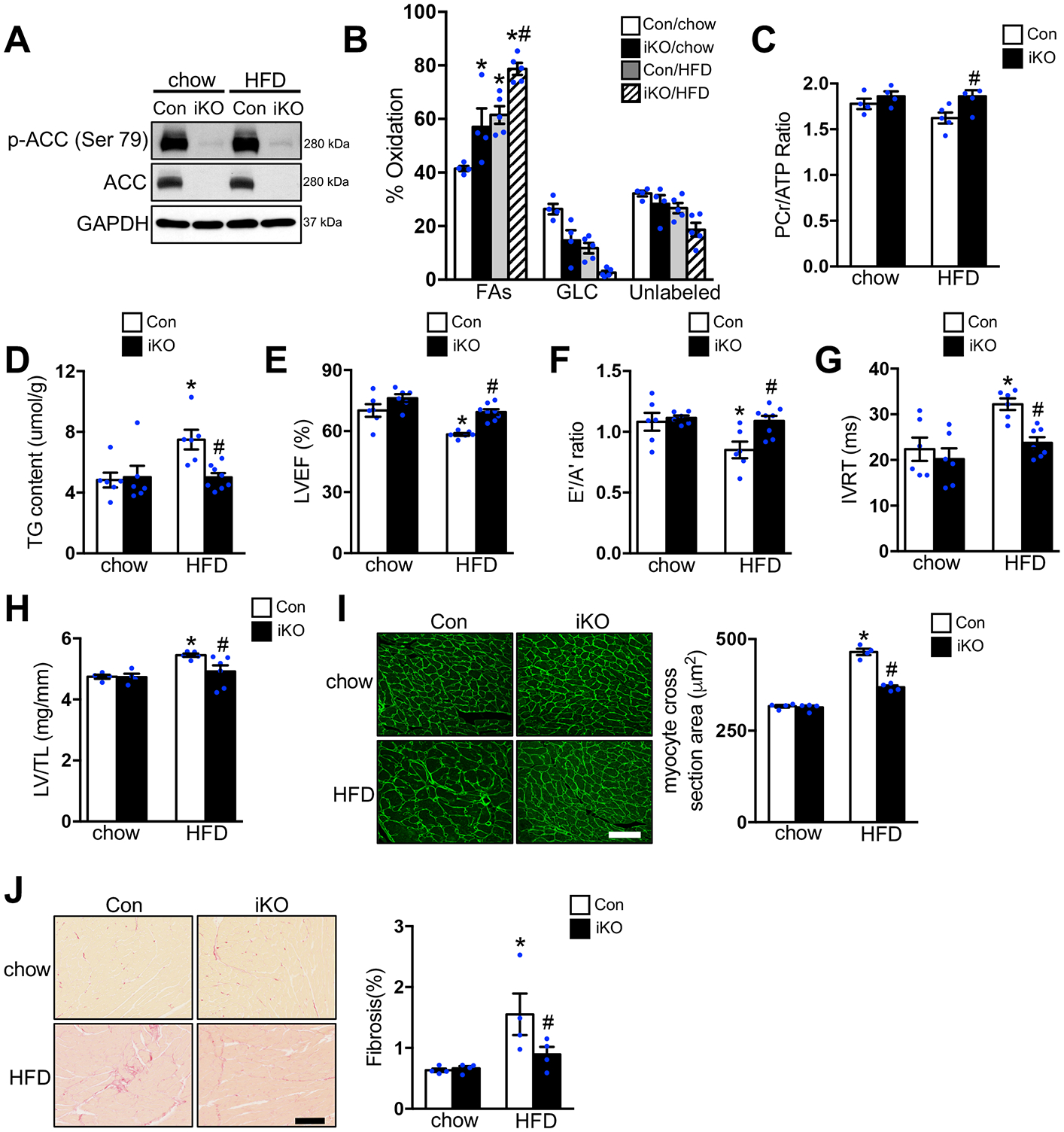
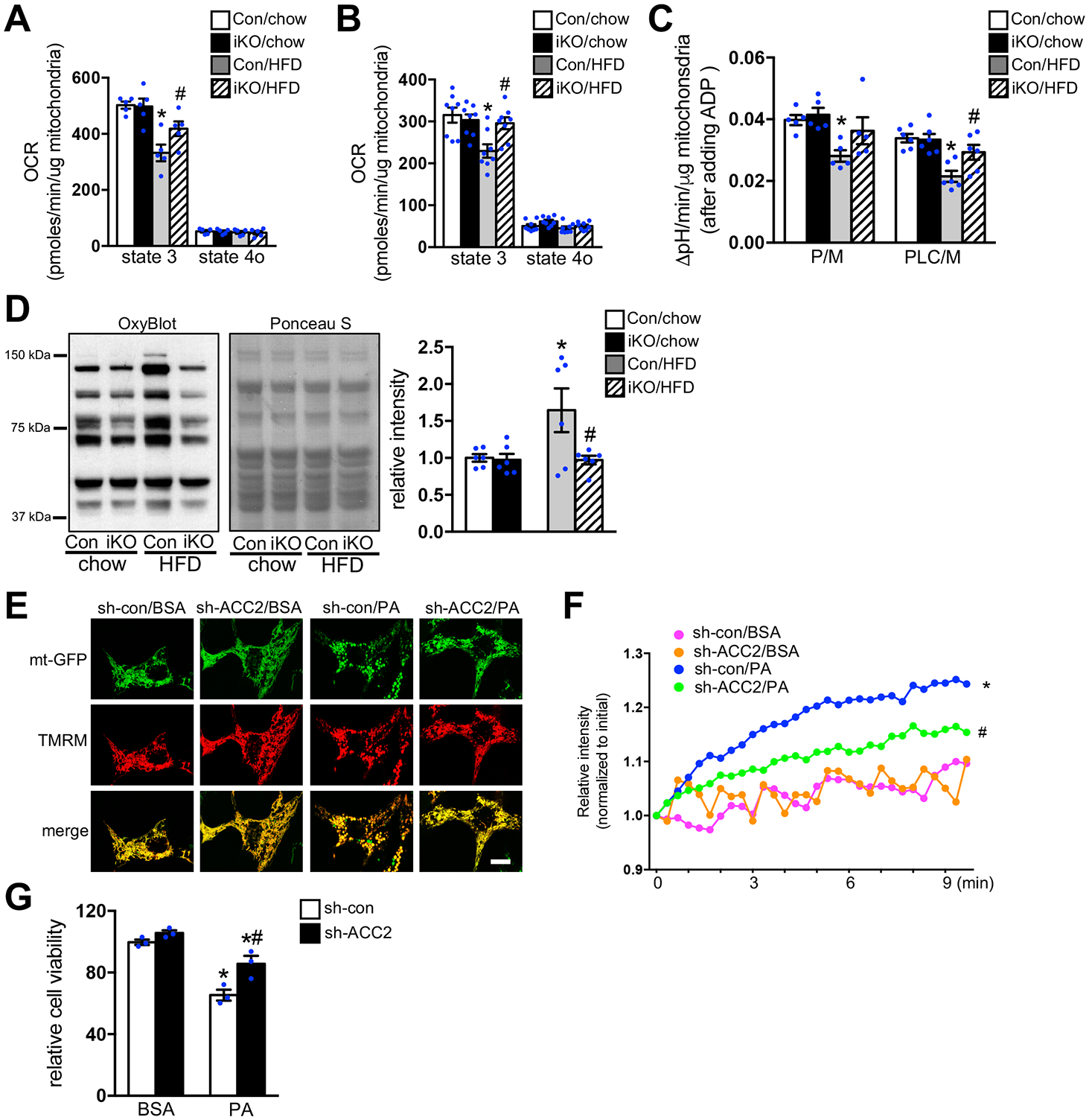
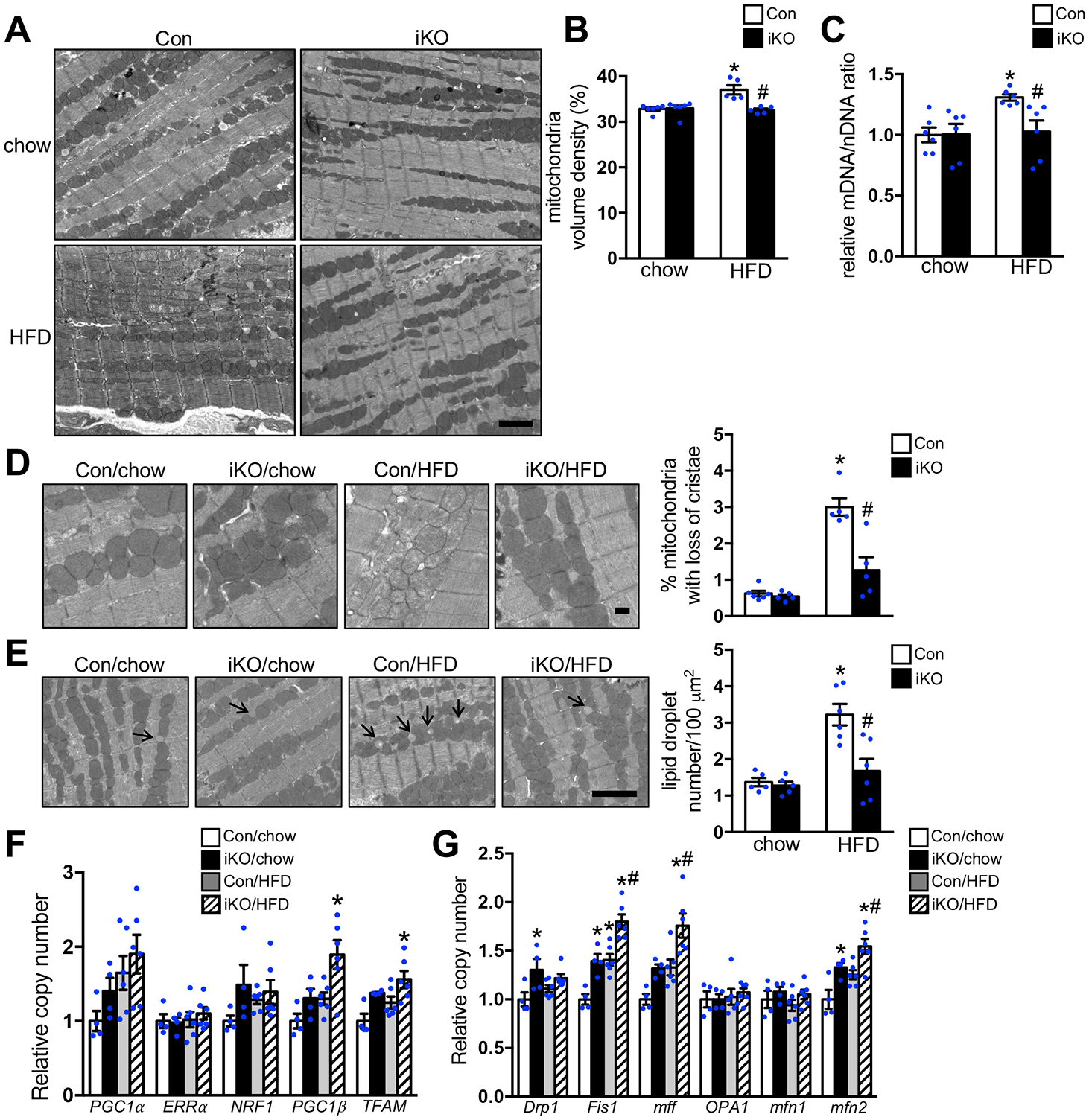
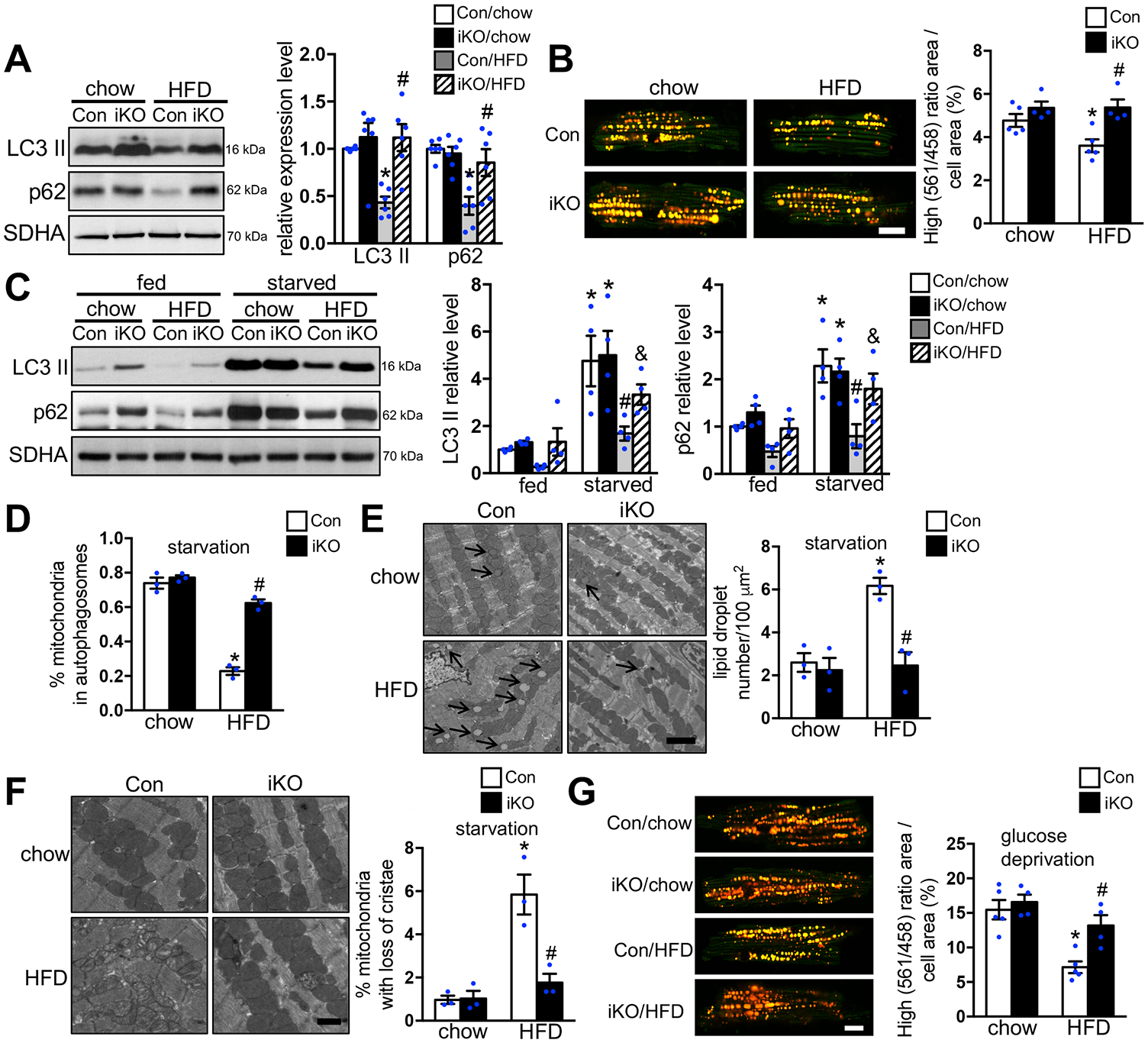
Background: Increased fatty acid oxidation (FAO) has long been considered a culprit in the development of obesity/diabetes mellitus-induced cardiomyopathy. However, enhancing cardiac FAO by removing the inhibitory mechanism of long-chain fatty acid transport into mitochondria via deletion of acetyl coenzyme A carboxylase 2 (ACC2) does not cause cardiomyopathy in nonobese mice, suggesting that high FAO is distinct from cardiac lipotoxicity. We hypothesize that cardiac pathology-associated obesity is attributable to the imbalance of fatty acid supply and oxidation. Thus, we here seek to determine whether further increasing FAO by inducing ACC2 deletion prevents obesity-induced cardiomyopathy, and if so, to elucidate the underlying mechanisms.
Methods: We induced high FAO in adult mouse hearts by cardiac-specific deletion of ACC2 using a tamoxifen-inducible model (ACC2 iKO). Control and ACC2 iKO mice were subjected to high-fat diet (HFD) feeding for 24 weeks to induce obesity. Cardiac function, mitochondria function, and mitophagy activity were examined.
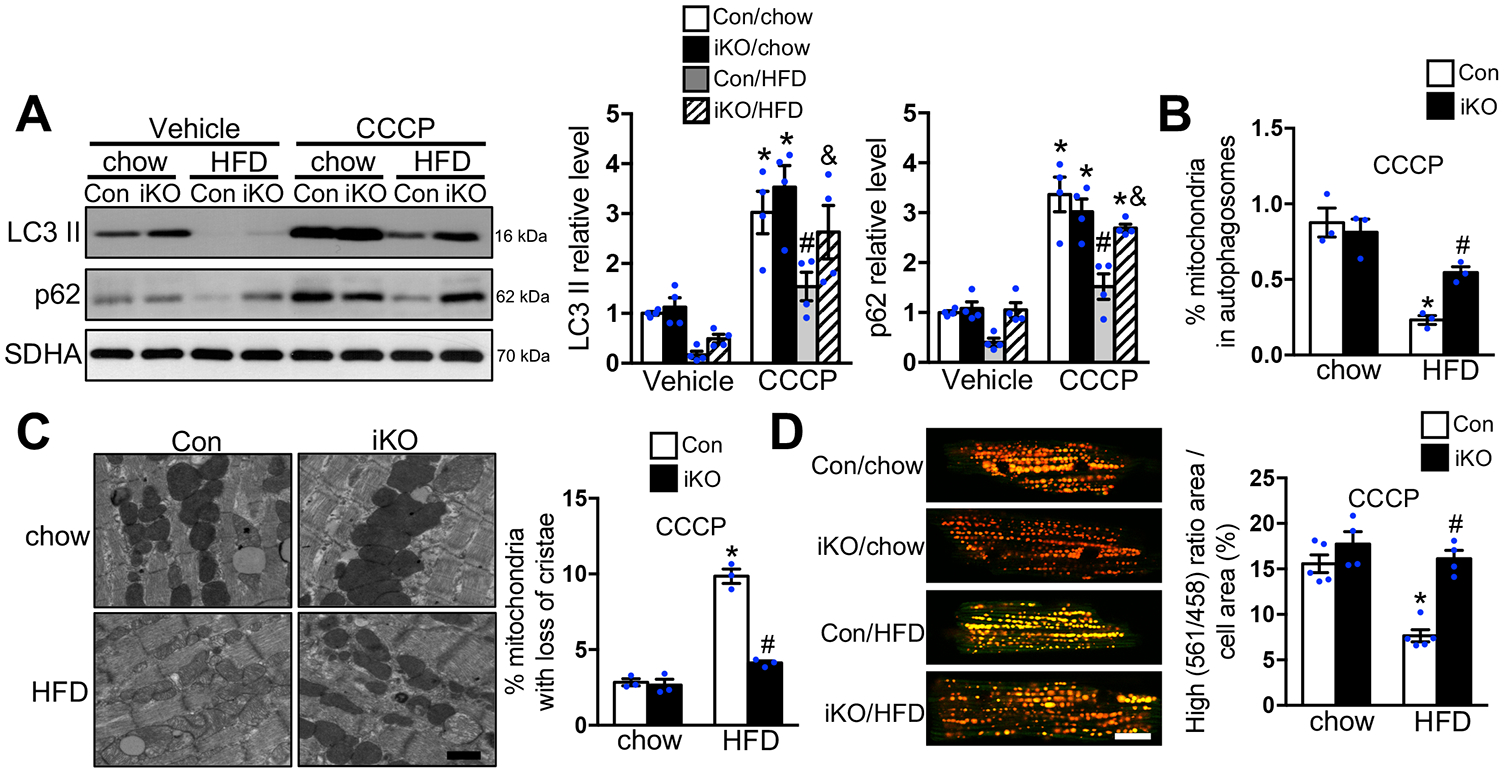
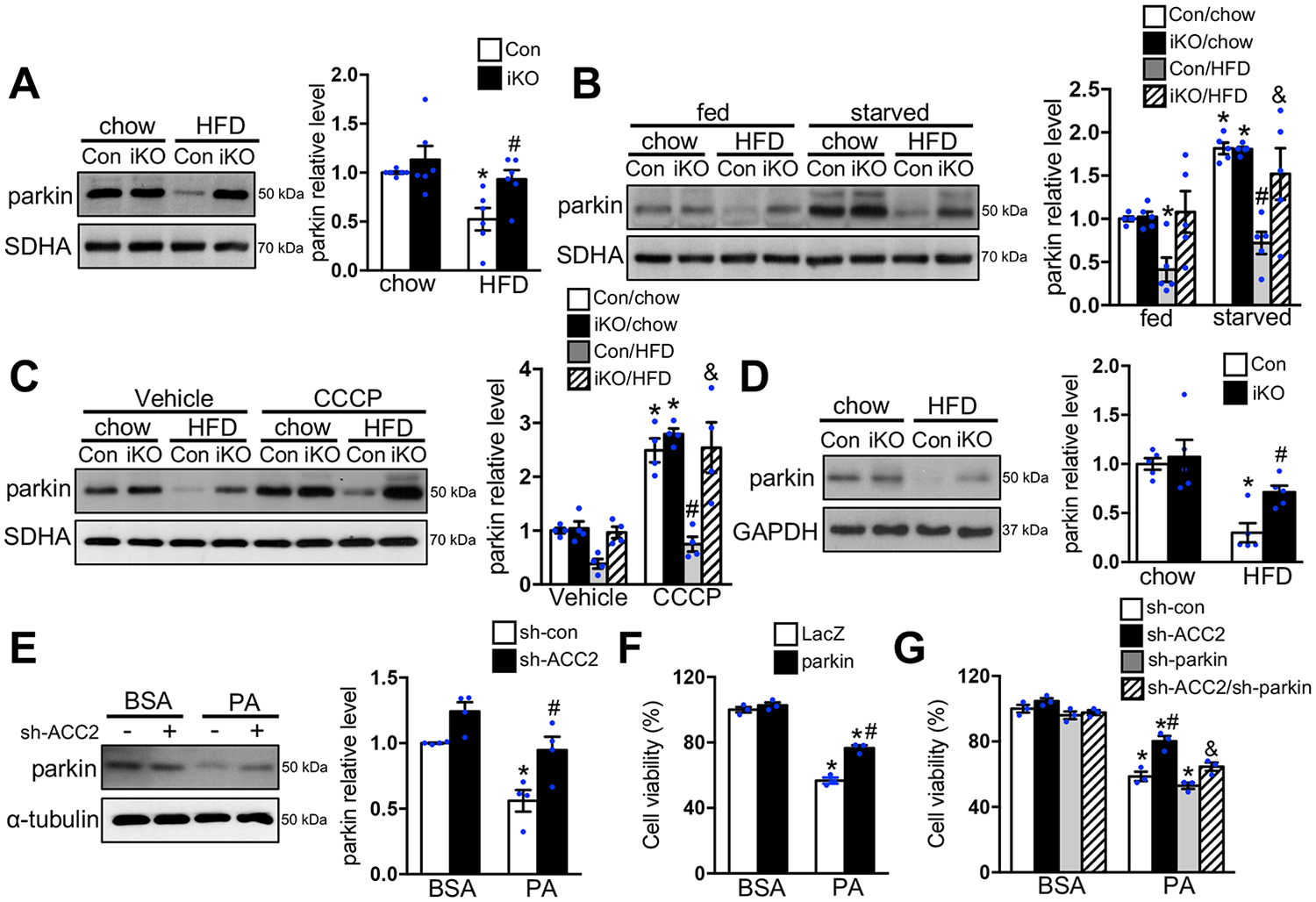
Results: Despite both control and ACC2 iKO mice exhibiting a similar obese phenotype, increasing FAO oxidation by deletion of ACC2 prevented HFD-induced cardiac dysfunction, pathological remodeling, and mitochondria dysfunction, as well. Similarly, increasing FAO by knockdown of ACC2 prevented palmitate-induced mitochondria dysfunction and cardiomyocyte death in vitro. Furthermore, HFD suppressed mitophagy activity and caused damaged mitochondria to accumulate in the heart, which was attenuated, in part, in the ACC2 iKO heart. Mechanistically, ACC2 iKO prevented HFD-induced downregulation of parkin. During stimulation for mitophagy, mitochondria-localized parkin was severely reduced in control HFD-fed mouse heart, which was restored, in part, in ACC2 iKO HFD-fed mice.
Conclusions: These data show that increasing cardiac FAO alone does not cause cardiac dysfunction, but protects against cardiomyopathy in chronically obese mice. The beneficial effect of enhancing cardiac FAO in HFD-induced obesity is mediated, in part, by the maintenance of mitochondria function through regulating parkin-mediated mitophagy. Our findings also suggest that targeting the parkin-dependent mitophagy pathway could be an effective strategy against the development of obesity-induced cardiomyopathy.
Keywords: cardiomyopathies; fatty acids; mitophagy; obesity.
Figures
References
-
- Fukushima A and Lopaschuk GD. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim Biophys Acta. 2016;1861:1525–1534. - PubMed
-
- Yang J, Sambandam N, Han X, Gross RW, Courtois M, Kovacs A, Febbraio M, Finck BN and Kelly DP. CD36 deficiency rescues lipotoxic cardiomyopathy. Circ Res. 2007;100:1208–1217. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
