

Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Intermolecular Interaction
- PMID: 32598850
- PMCID: PMC7710120
- DOI: 10.1021/acs.chemrev.9b00813
Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Intermolecular Interaction
Erratum in
-
Correction to Vibrational Spectroscopic Map, Vibrational Spectroscopy, and Intermolecular Interaction.Chem Rev. 2021 Nov 10;121(21):13698. doi: 10.1021/acs.chemrev.1c00758. Epub 2021 Oct 28. Chem Rev. 2021. PMID: 34709802 No abstract available.
Abstract
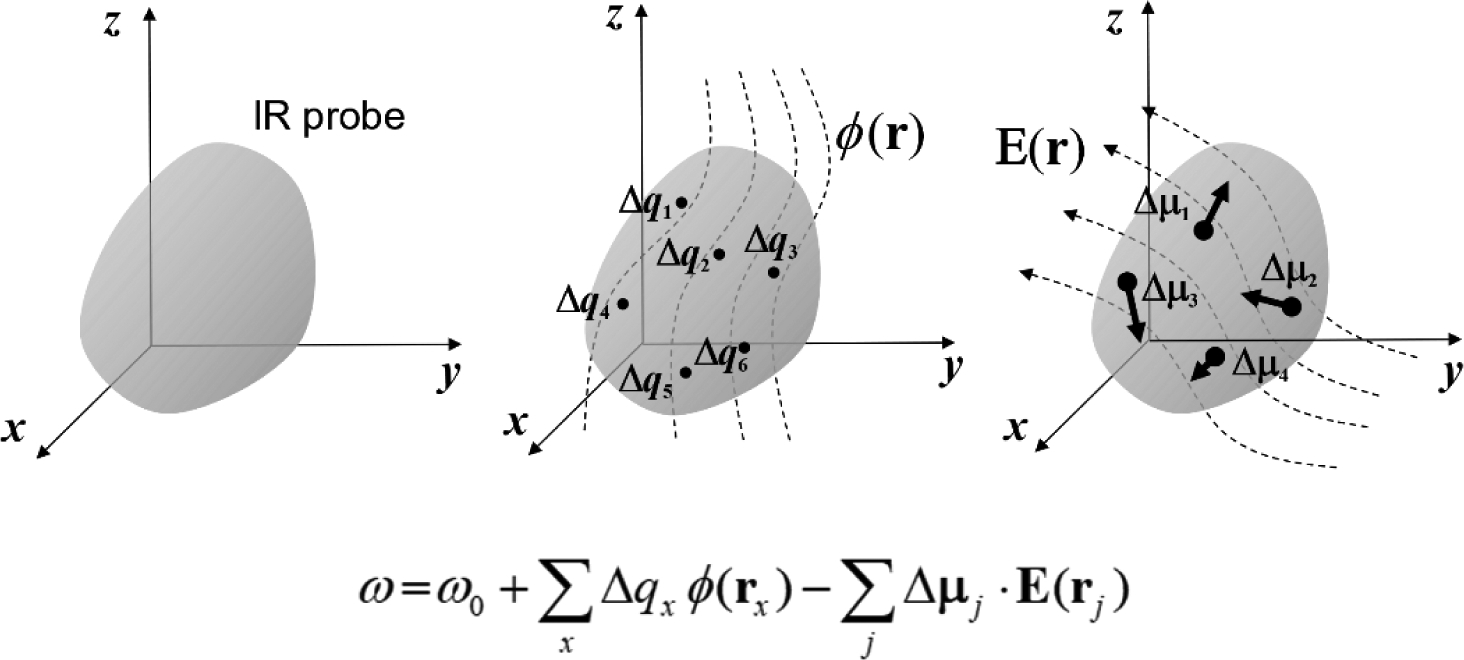
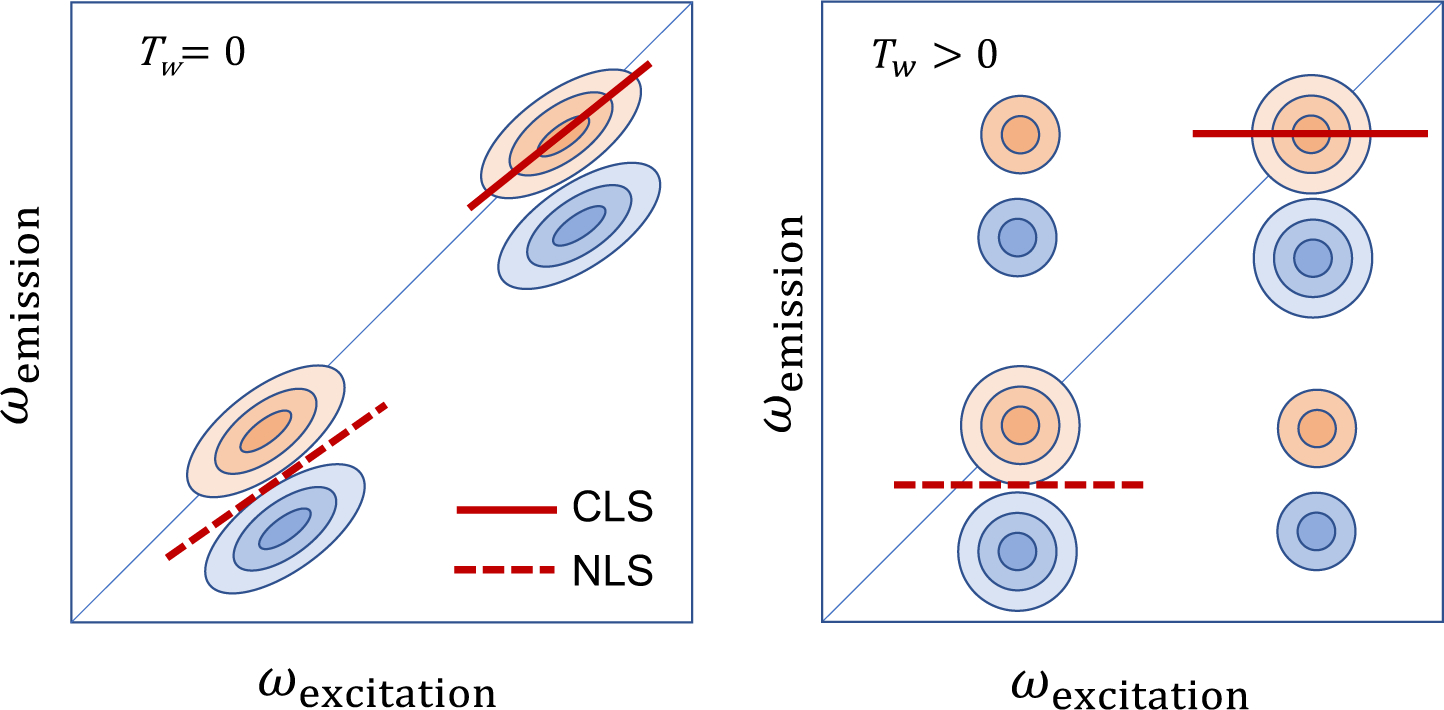
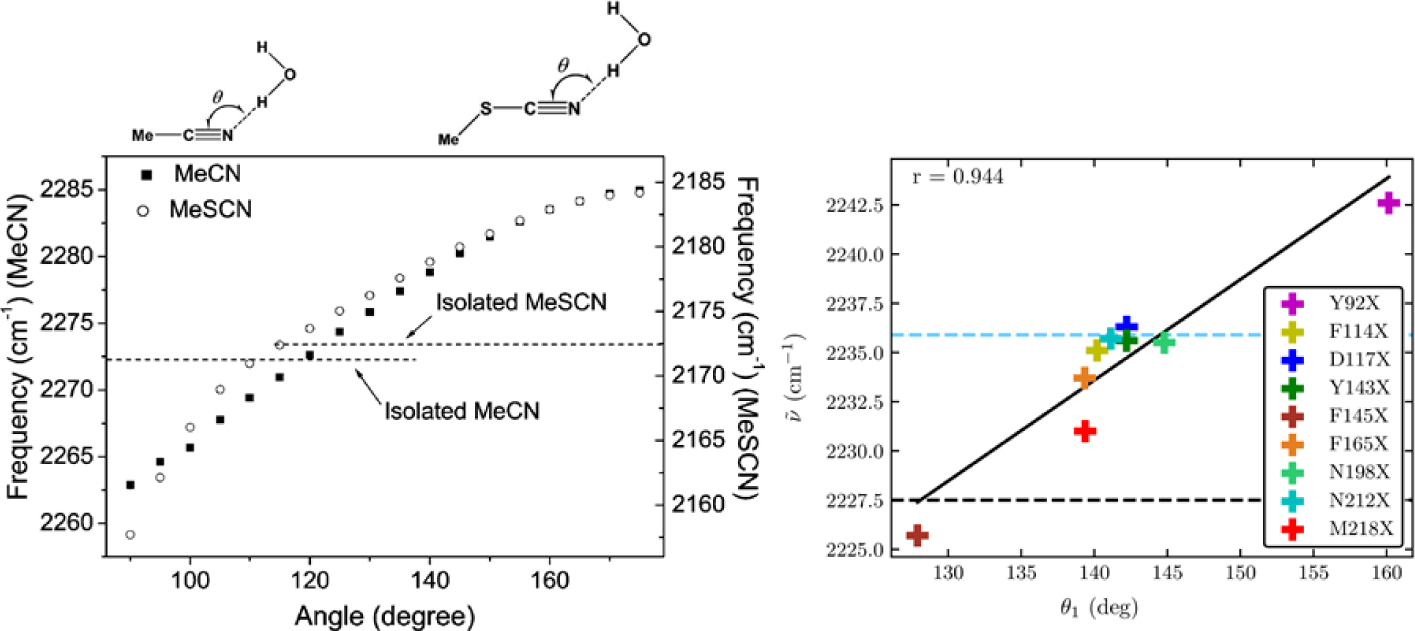
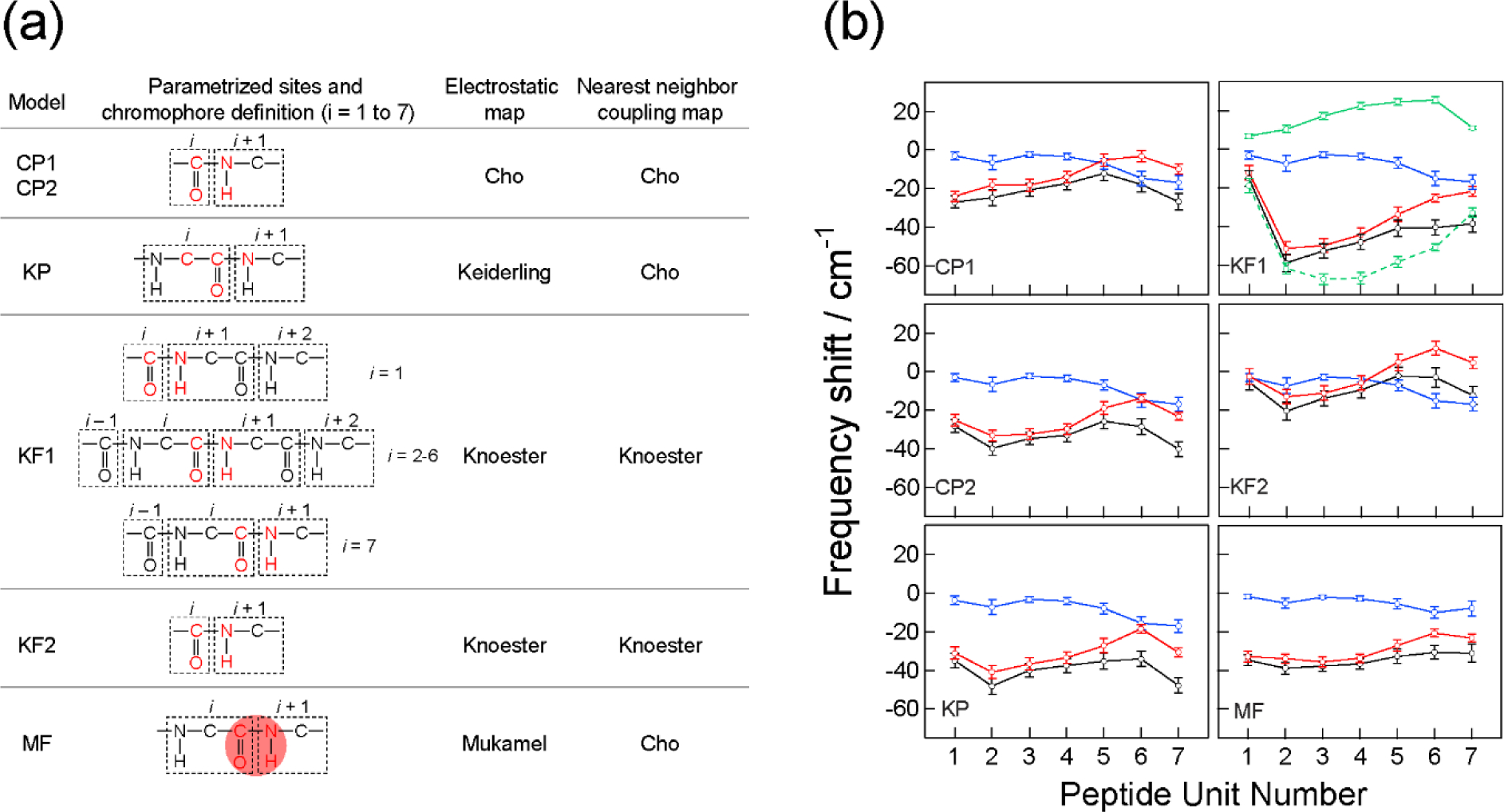
Vibrational spectroscopy is an essential tool in chemical analyses, biological assays, and studies of functional materials. Over the past decade, various coherent nonlinear vibrational spectroscopic techniques have been developed and enabled researchers to study time-correlations of the fluctuating frequencies that are directly related to solute-solvent dynamics, dynamical changes in molecular conformations and local electrostatic environments, chemical and biochemical reactions, protein structural dynamics and functions, characteristic processes of functional materials, and so on. In order to gain incisive and quantitative information on the local electrostatic environment, molecular conformation, protein structure and interprotein contacts, ligand binding kinetics, and electric and optical properties of functional materials, a variety of vibrational probes have been developed and site-specifically incorporated into molecular, biological, and material systems for time-resolved vibrational spectroscopic investigation. However, still, an all-encompassing theory that describes the vibrational solvatochromism, electrochromism, and dynamic fluctuation of vibrational frequencies has not been completely established mainly due to the intrinsic complexity of intermolecular interactions in condensed phases. In particular, the amount of data obtained from the linear and nonlinear vibrational spectroscopic experiments has been rapidly increasing, but the lack of a quantitative method to interpret these measurements has been one major obstacle in broadening the applications of these methods. Among various theoretical models, one of the most successful approaches is a semiempirical model generally referred to as the vibrational spectroscopic map that is based on a rigorous theory of intermolecular interactions. Recently, genetic algorithm, neural network, and machine learning approaches have been applied to the development of vibrational solvatochromism theory. In this review, we provide comprehensive descriptions of the theoretical foundation and various examples showing its extraordinary successes in the interpretations of experimental observations. In addition, a brief introduction to a newly created repository Web site (http://frequencymap.org) for vibrational spectroscopic maps is presented. We anticipate that a combination of the vibrational frequency map approach and state-of-the-art multidimensional vibrational spectroscopy will be one of the most fruitful ways to study the structure and dynamics of chemical, biological, and functional molecular systems in the future.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
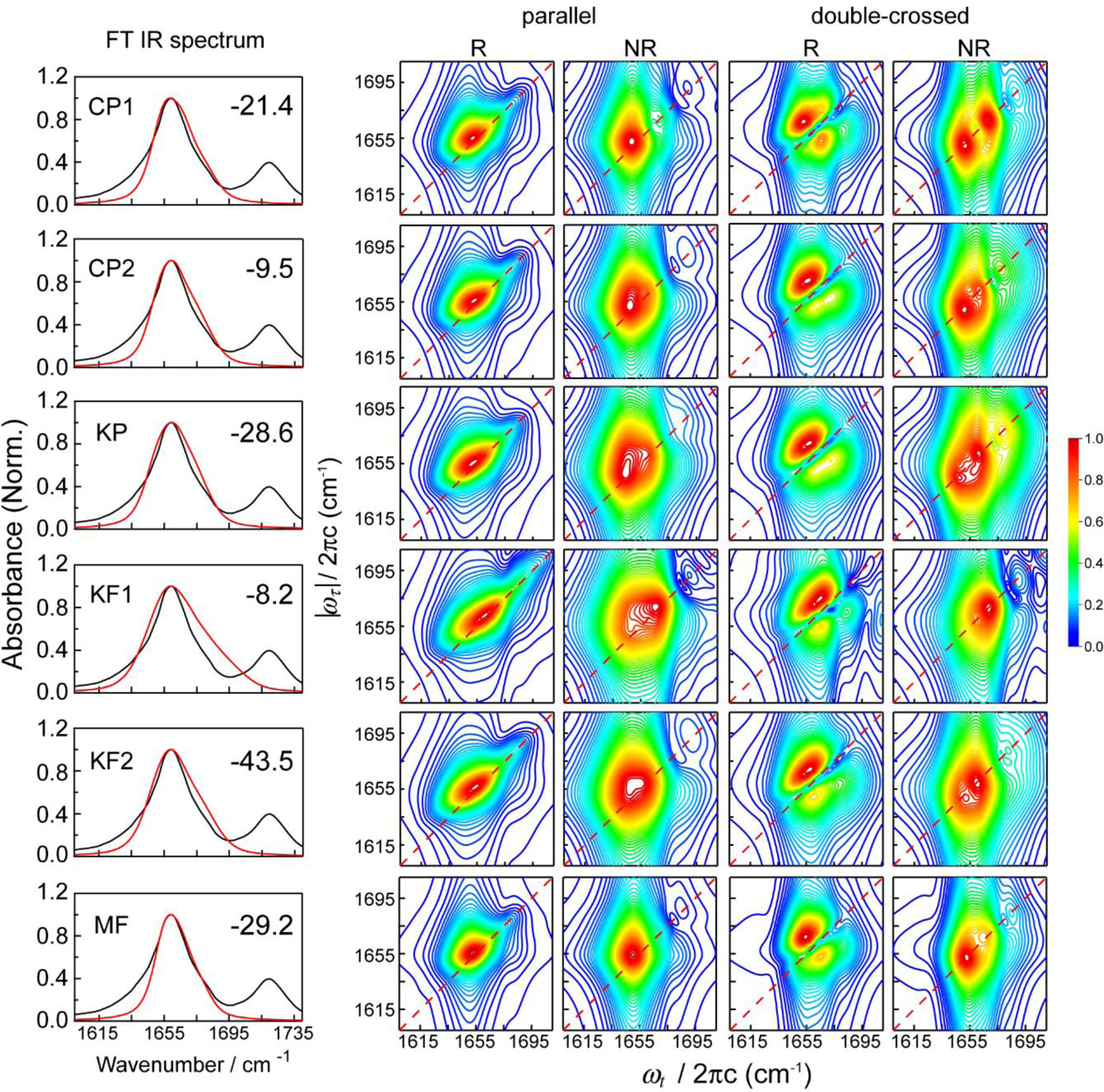
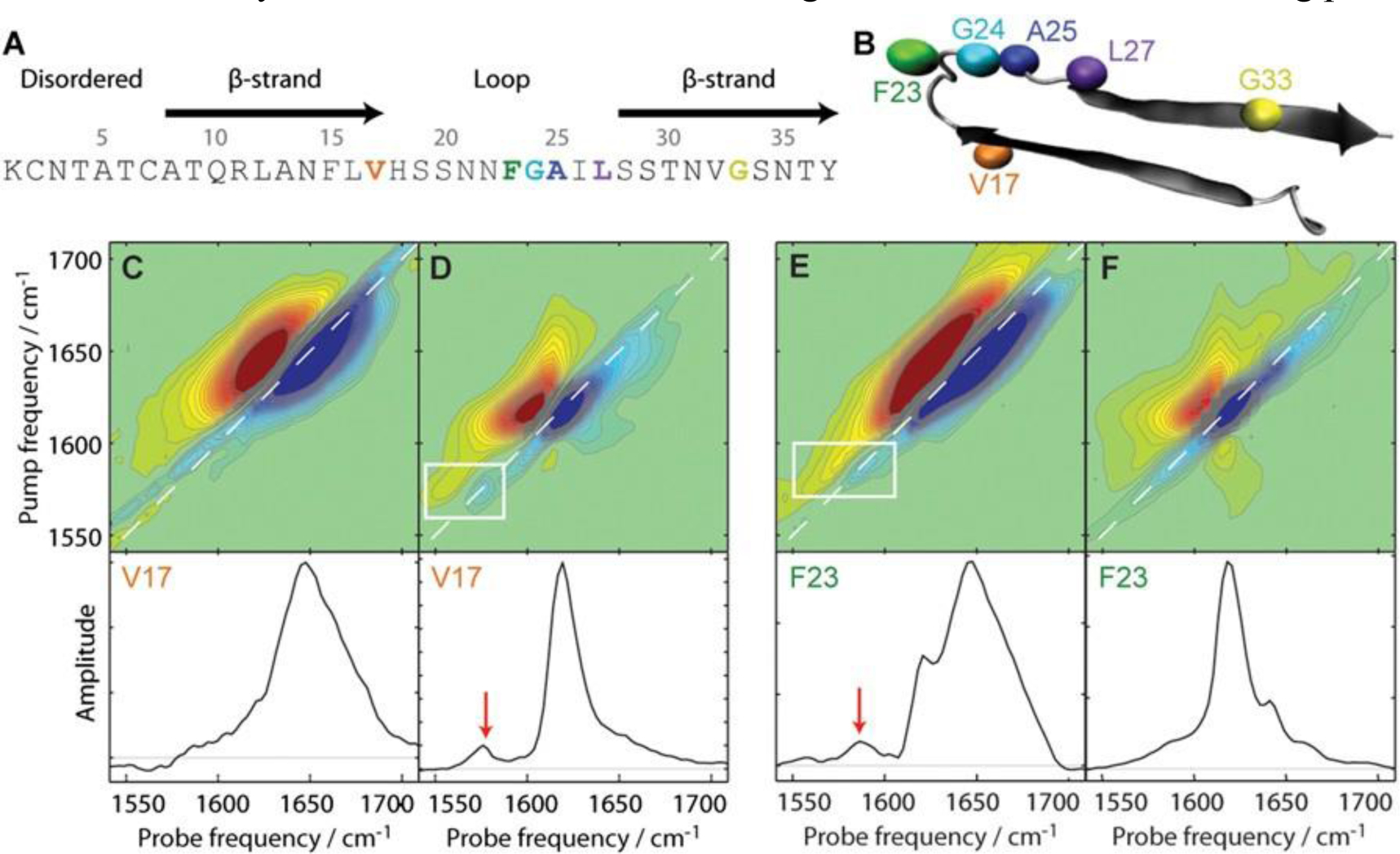
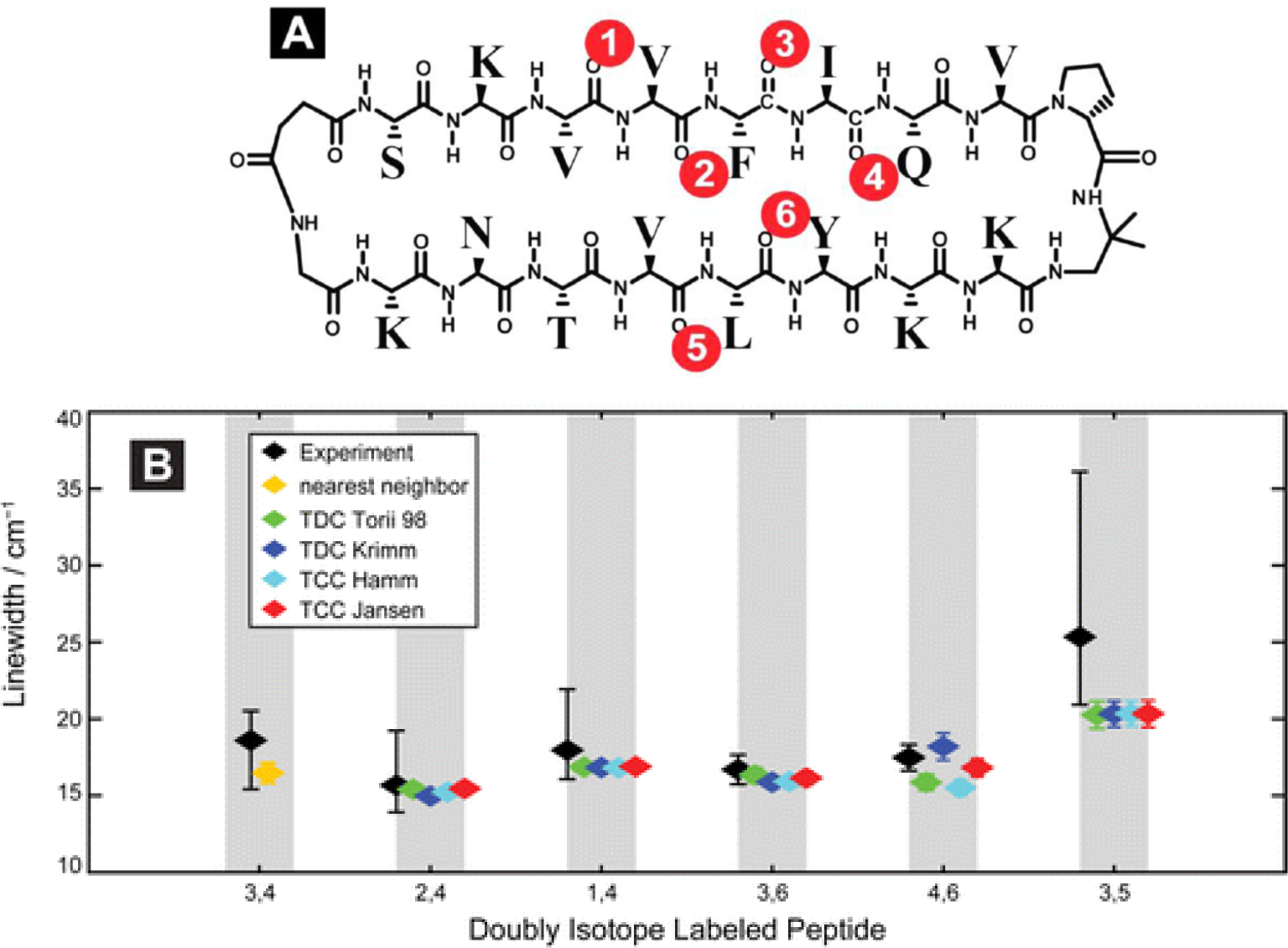
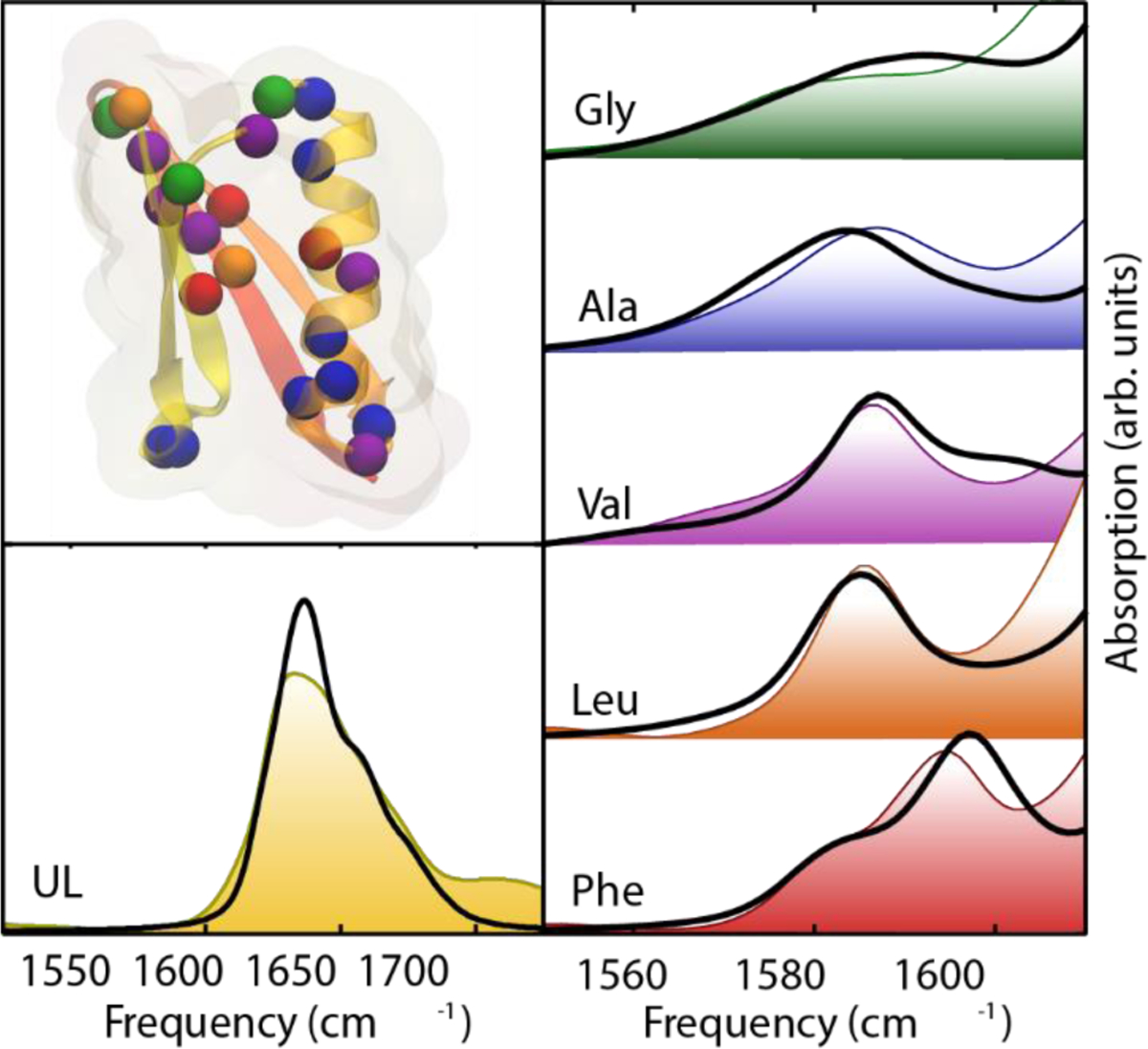
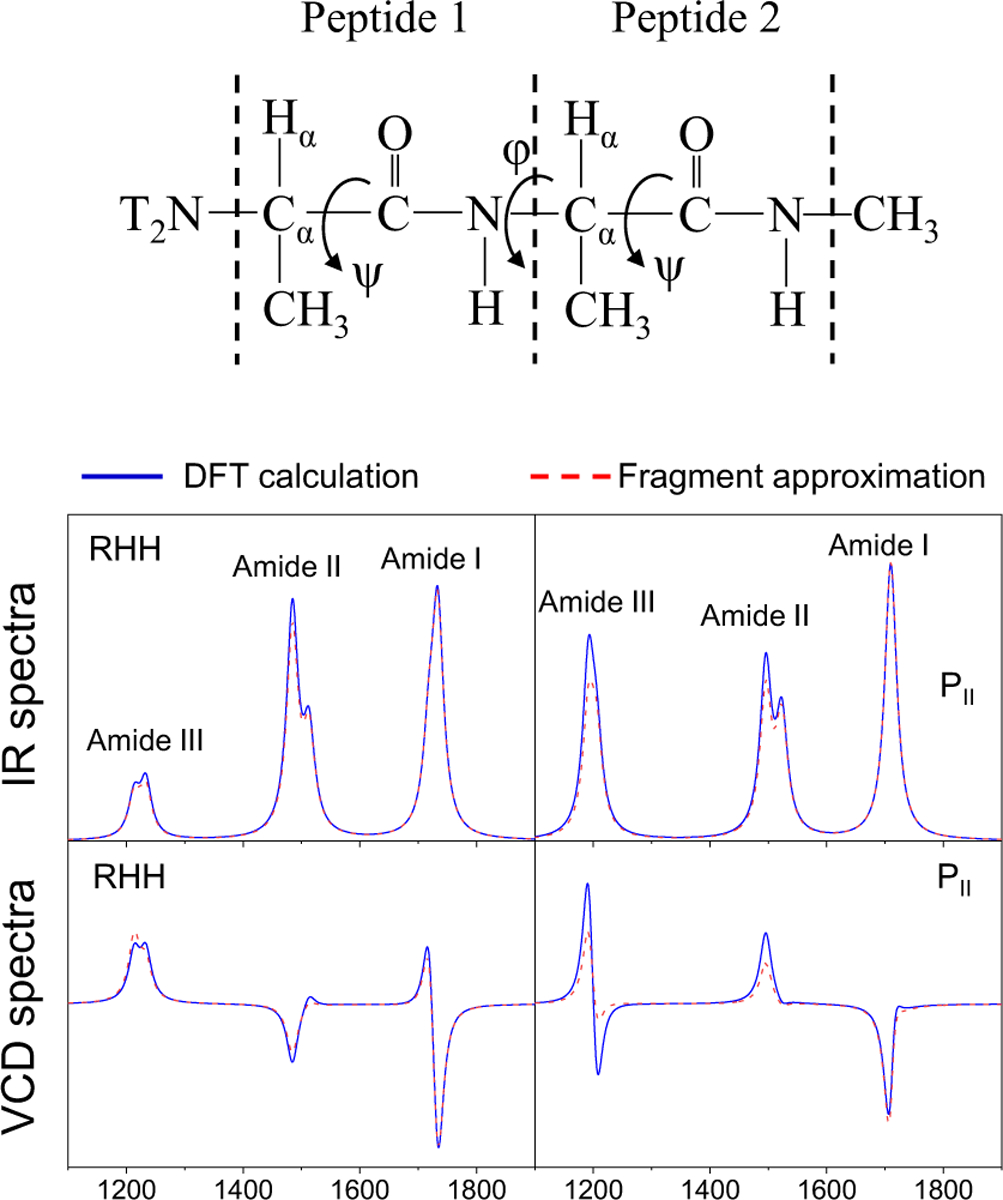
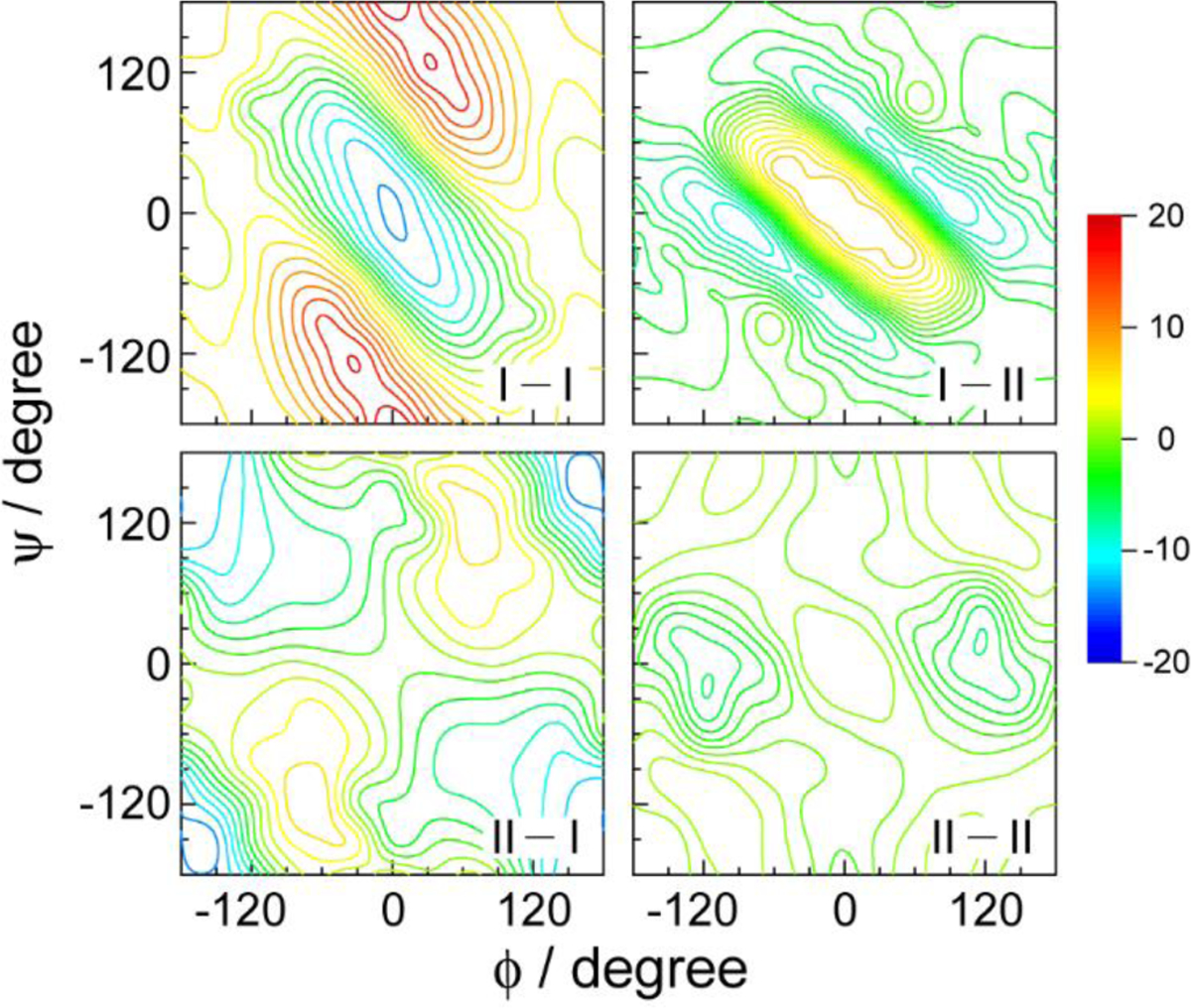
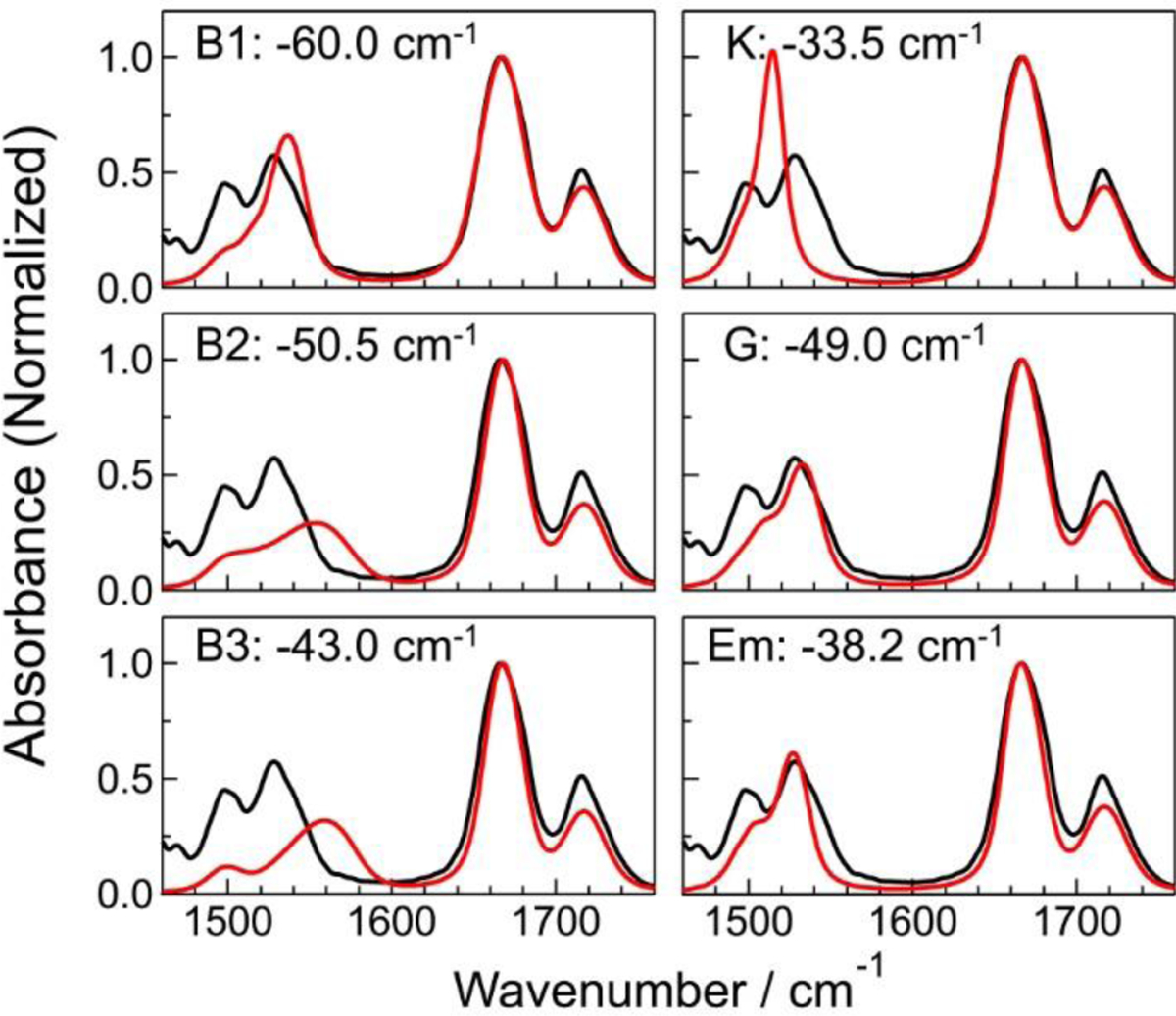
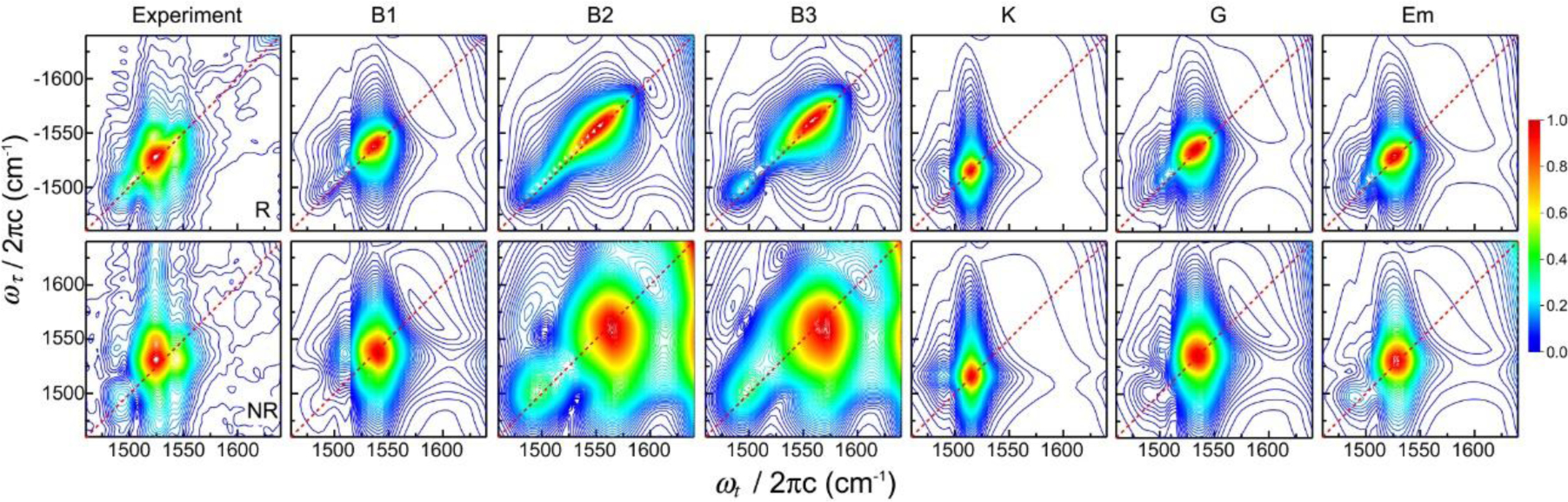
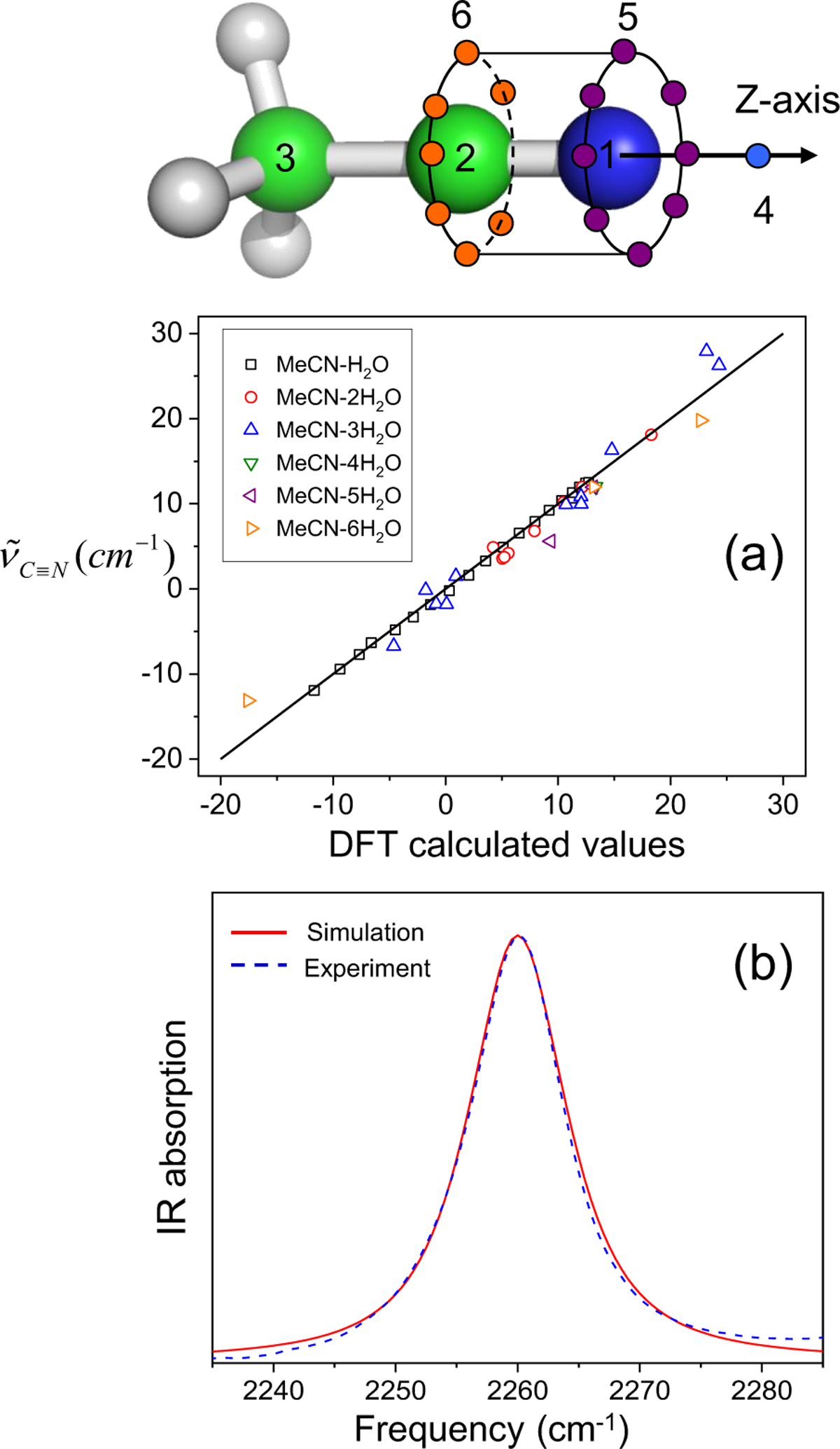
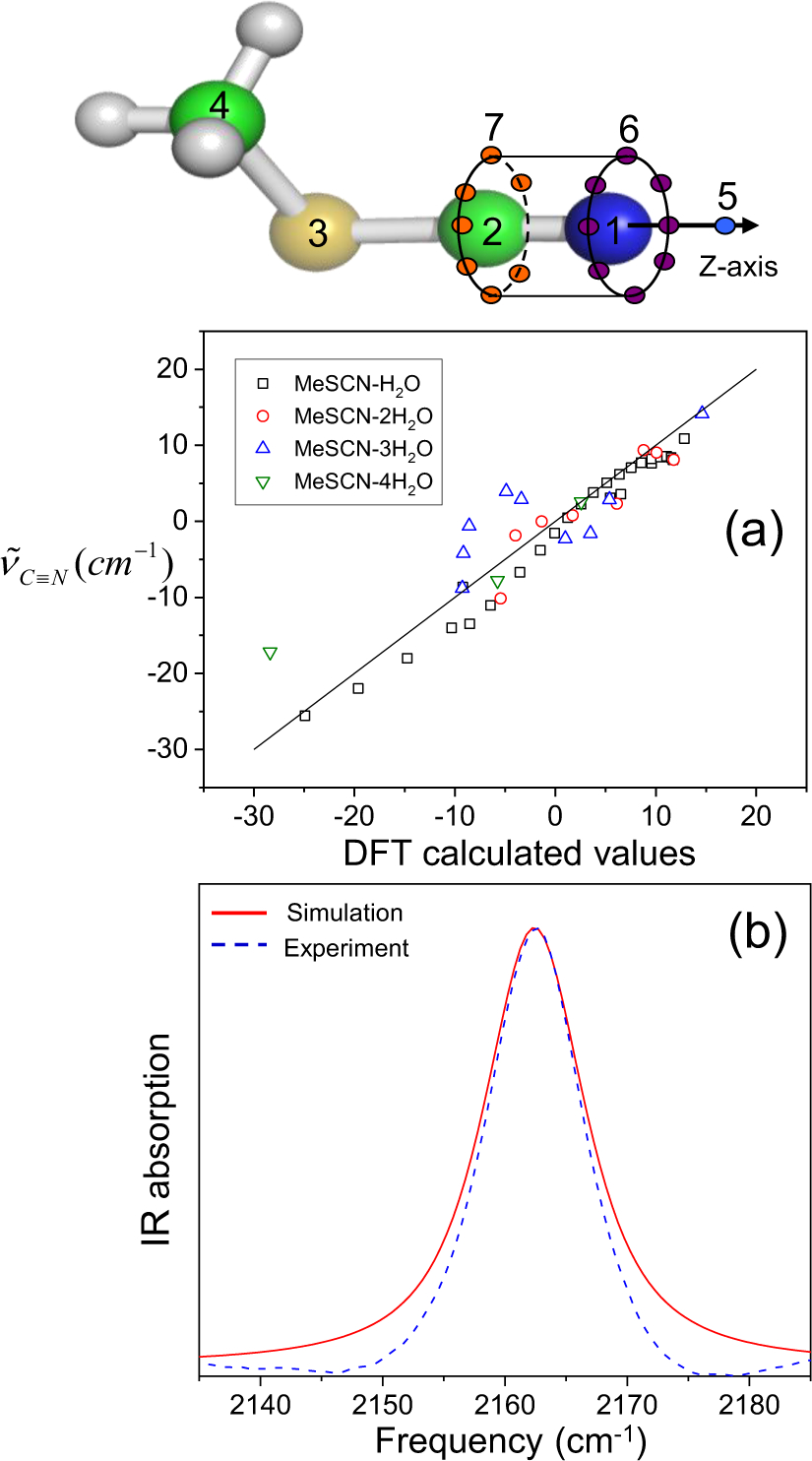
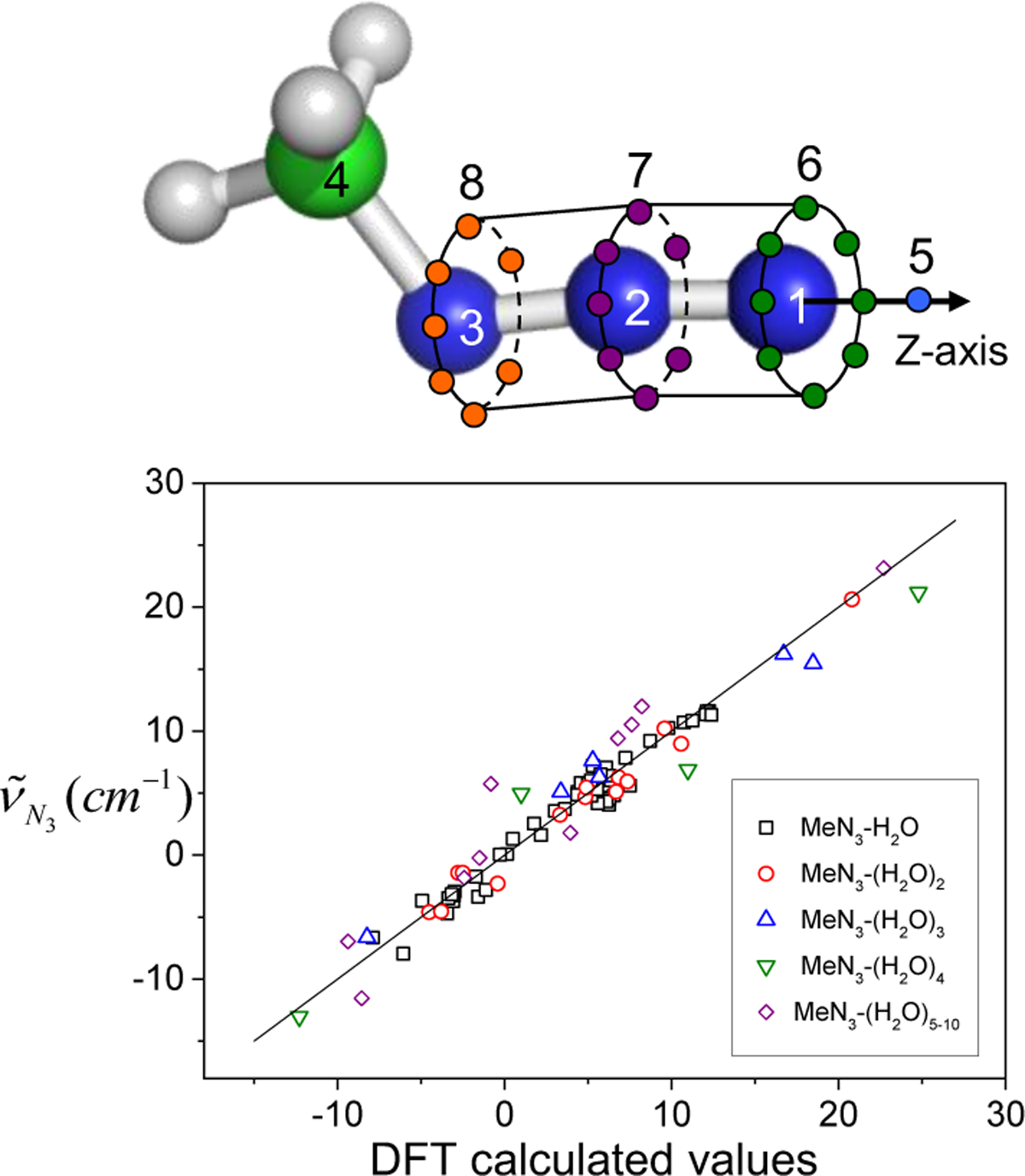
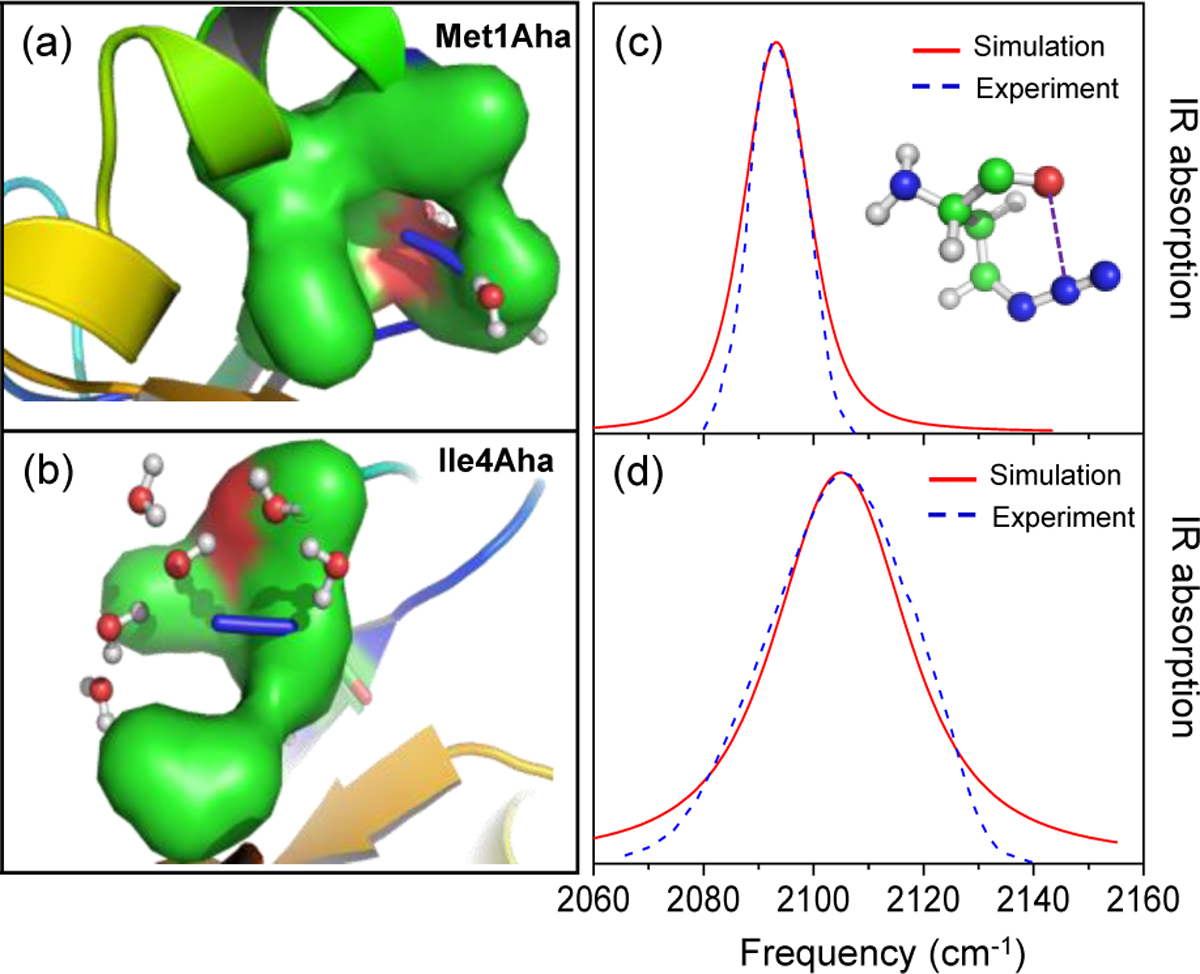
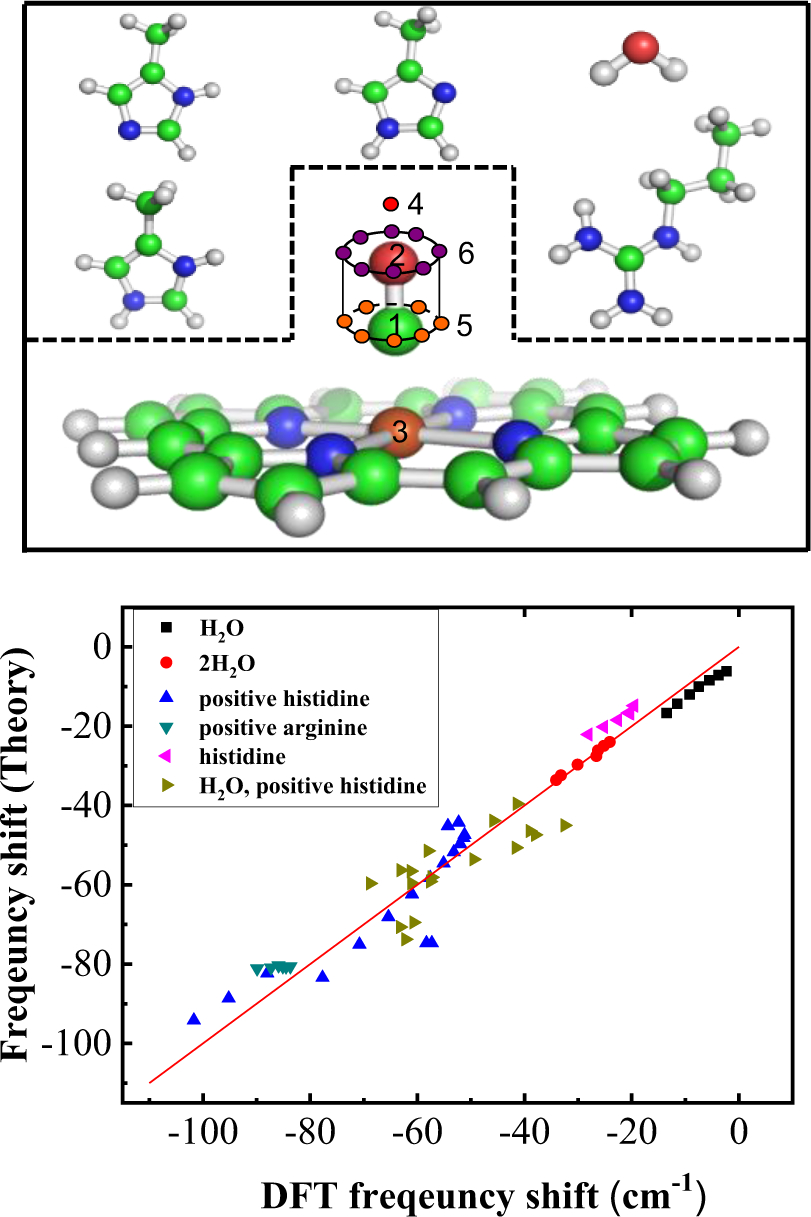
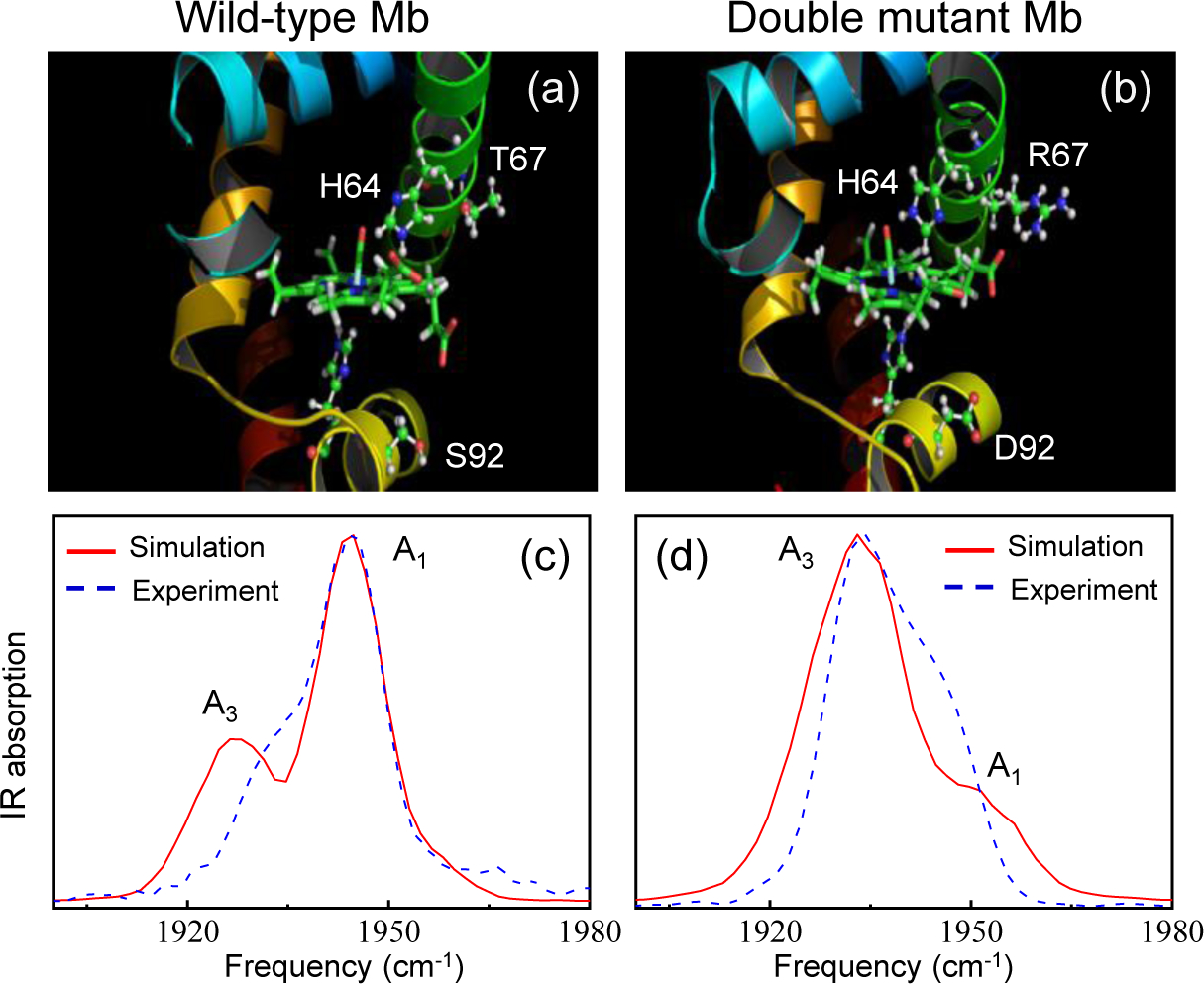
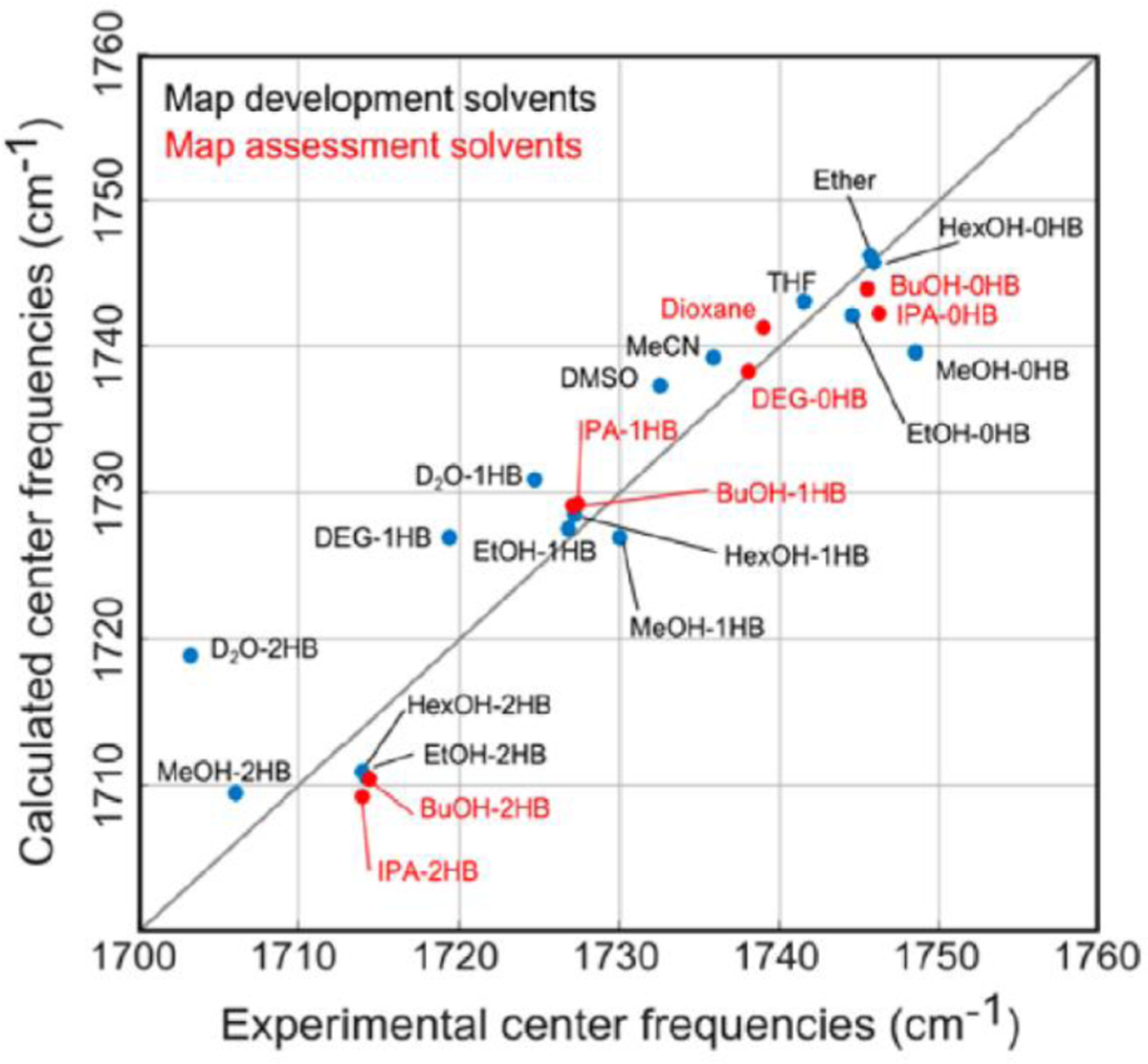
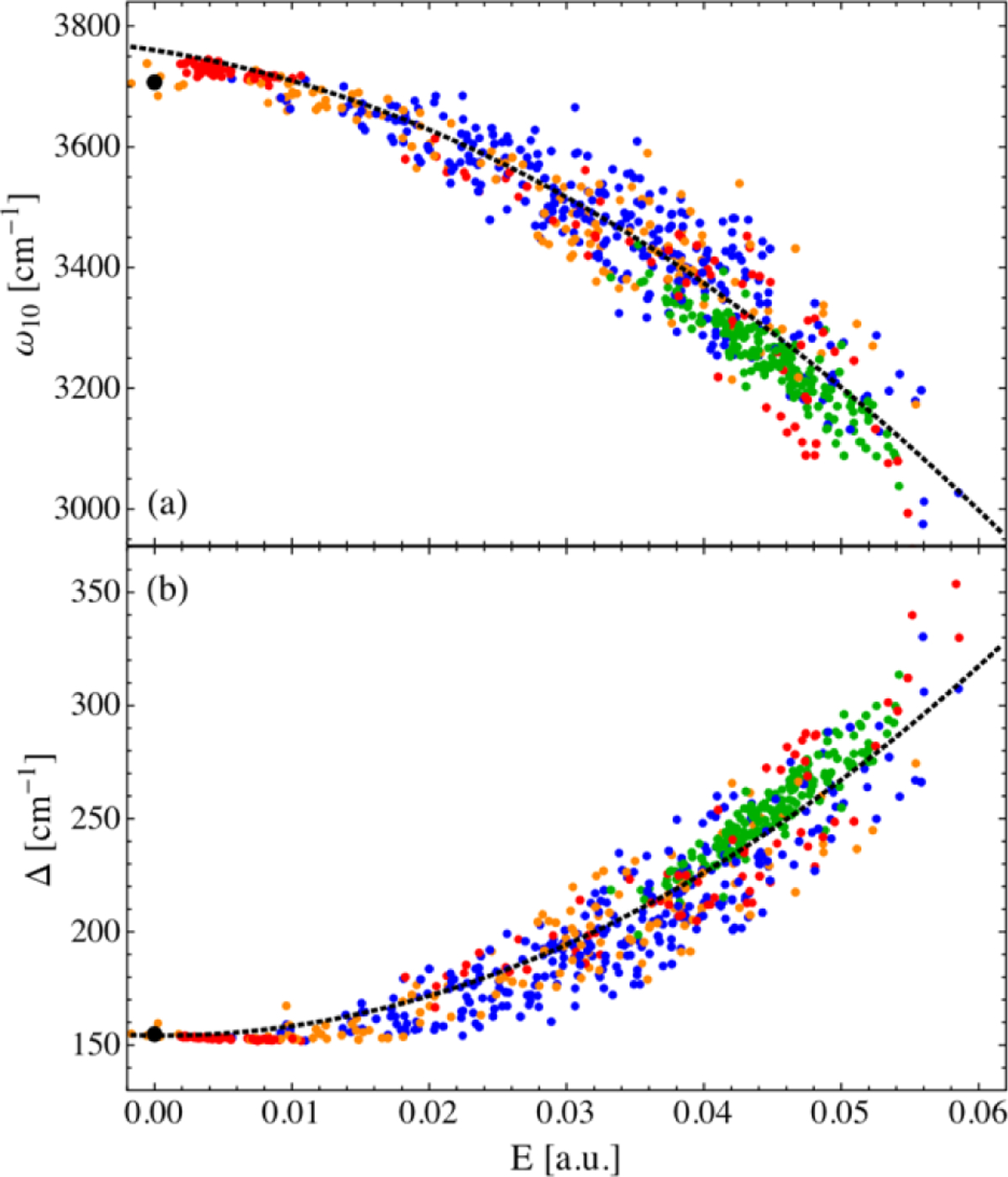
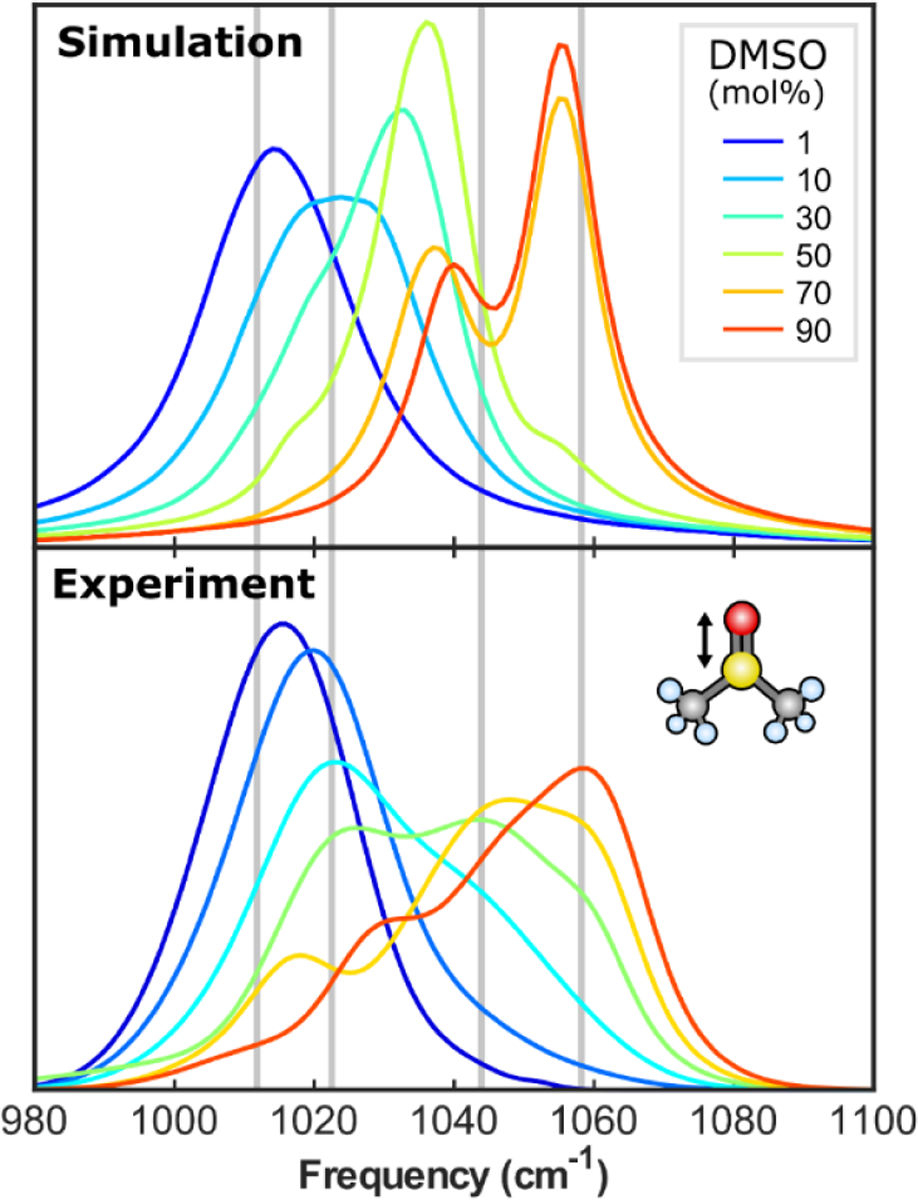
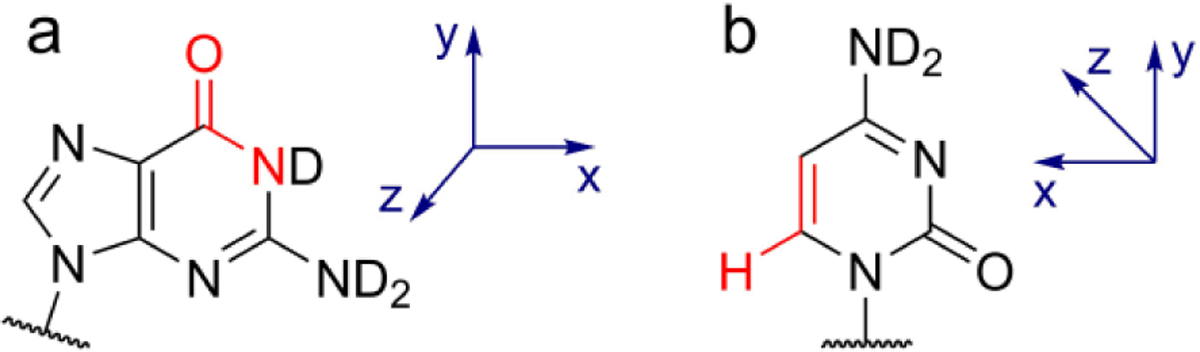
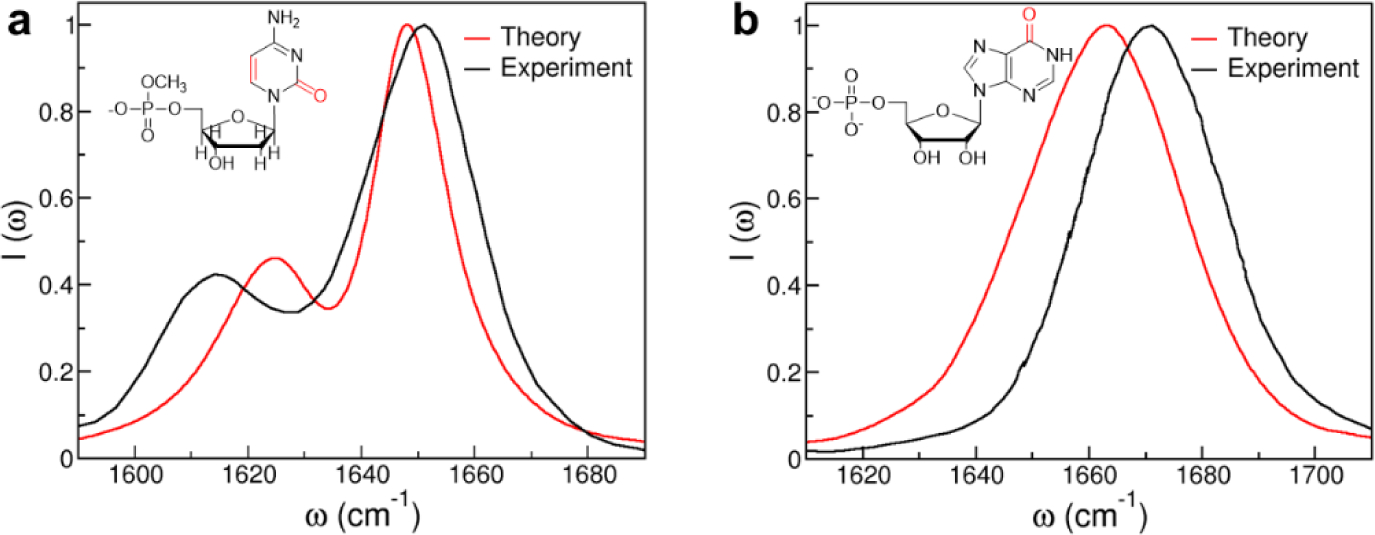
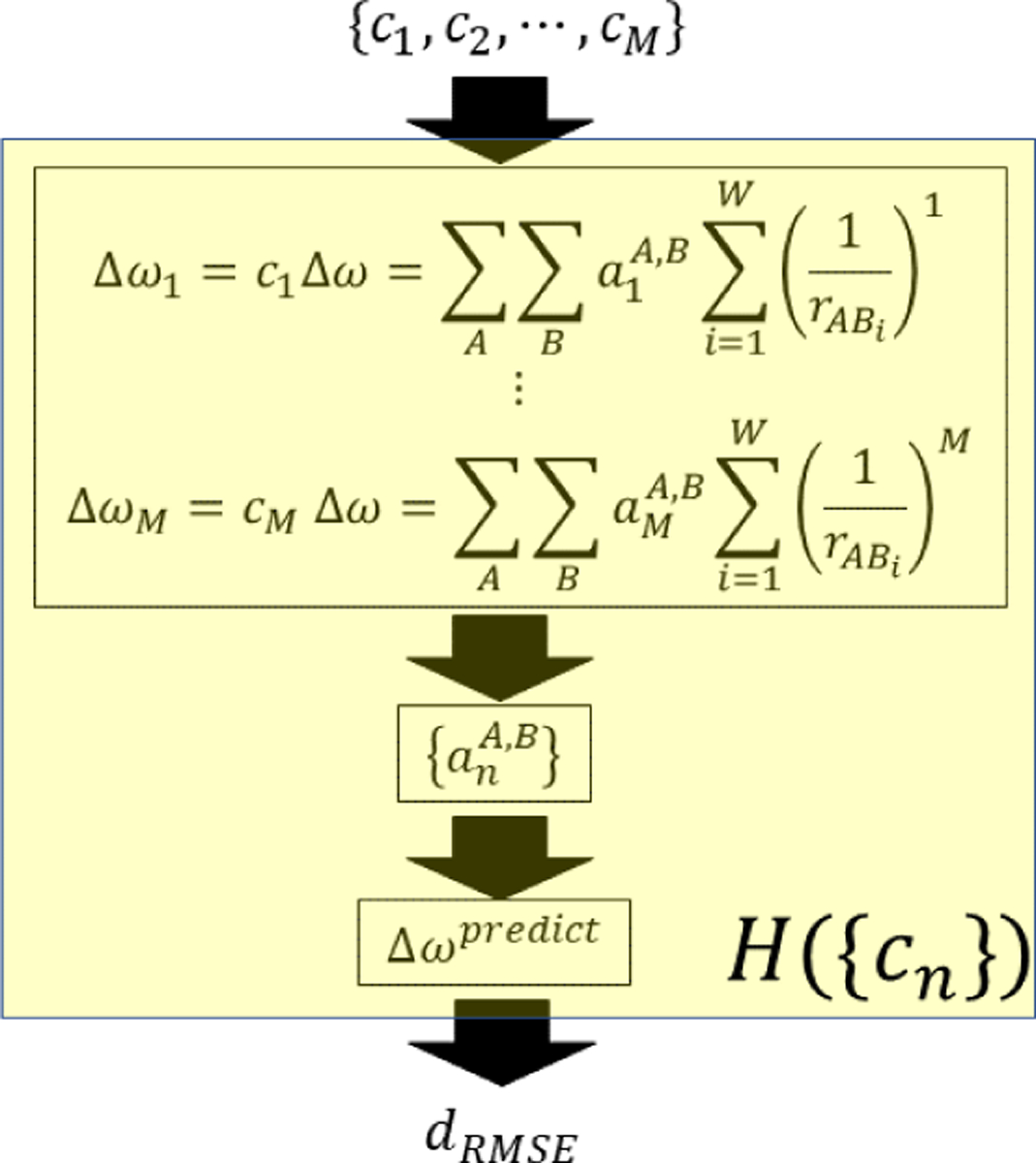
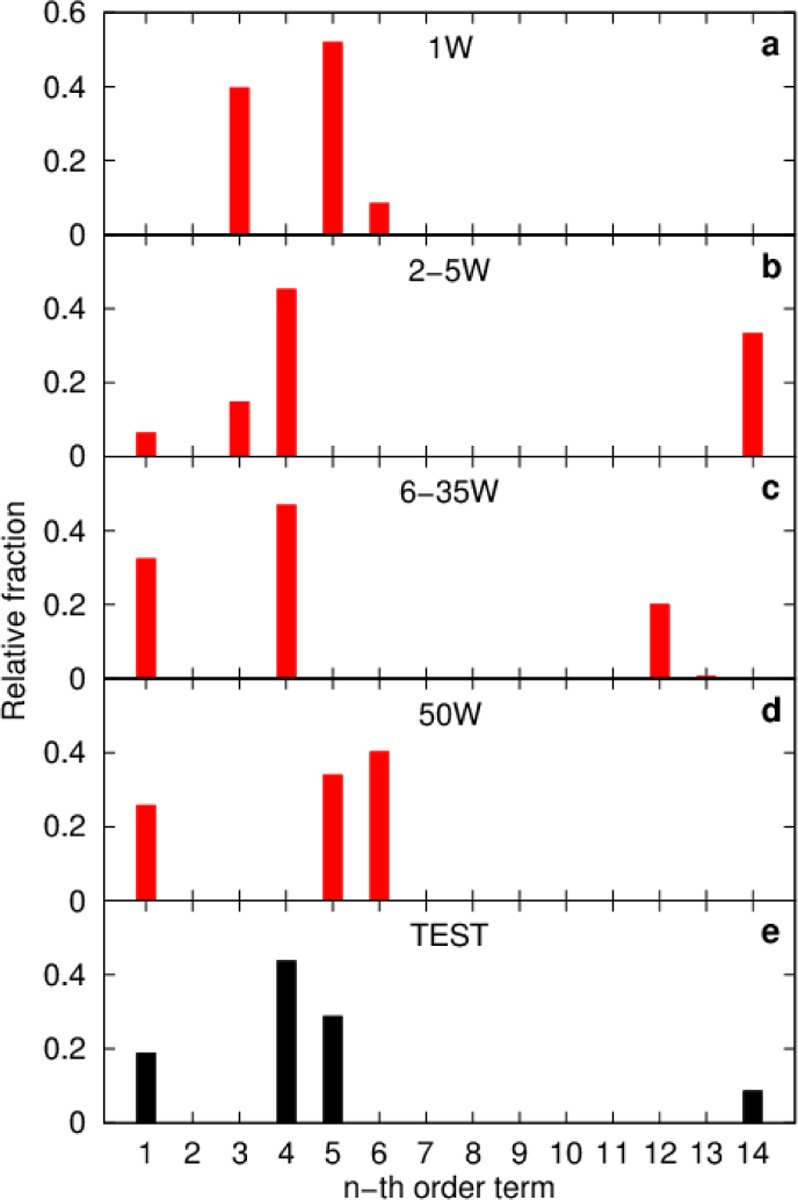
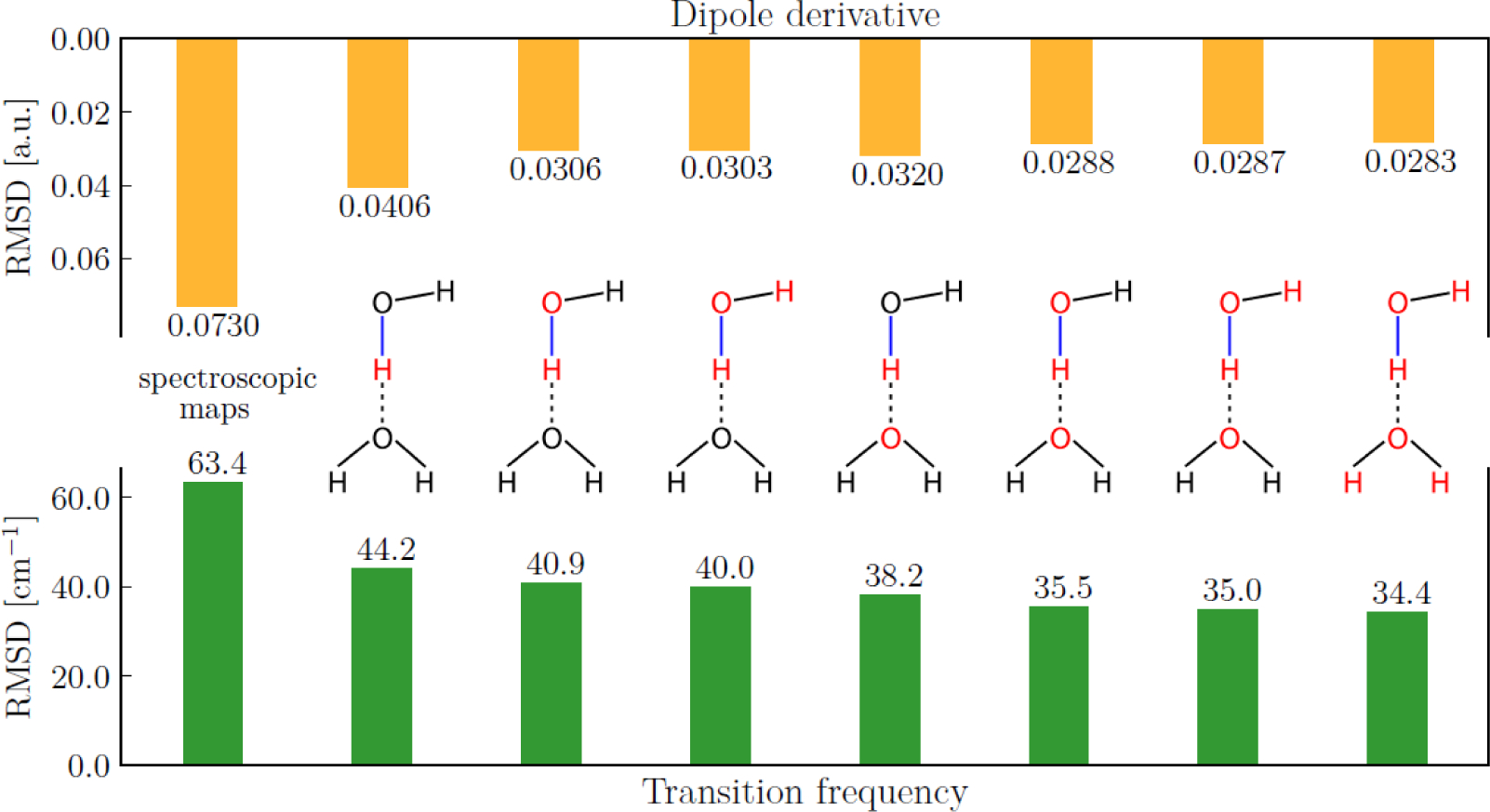
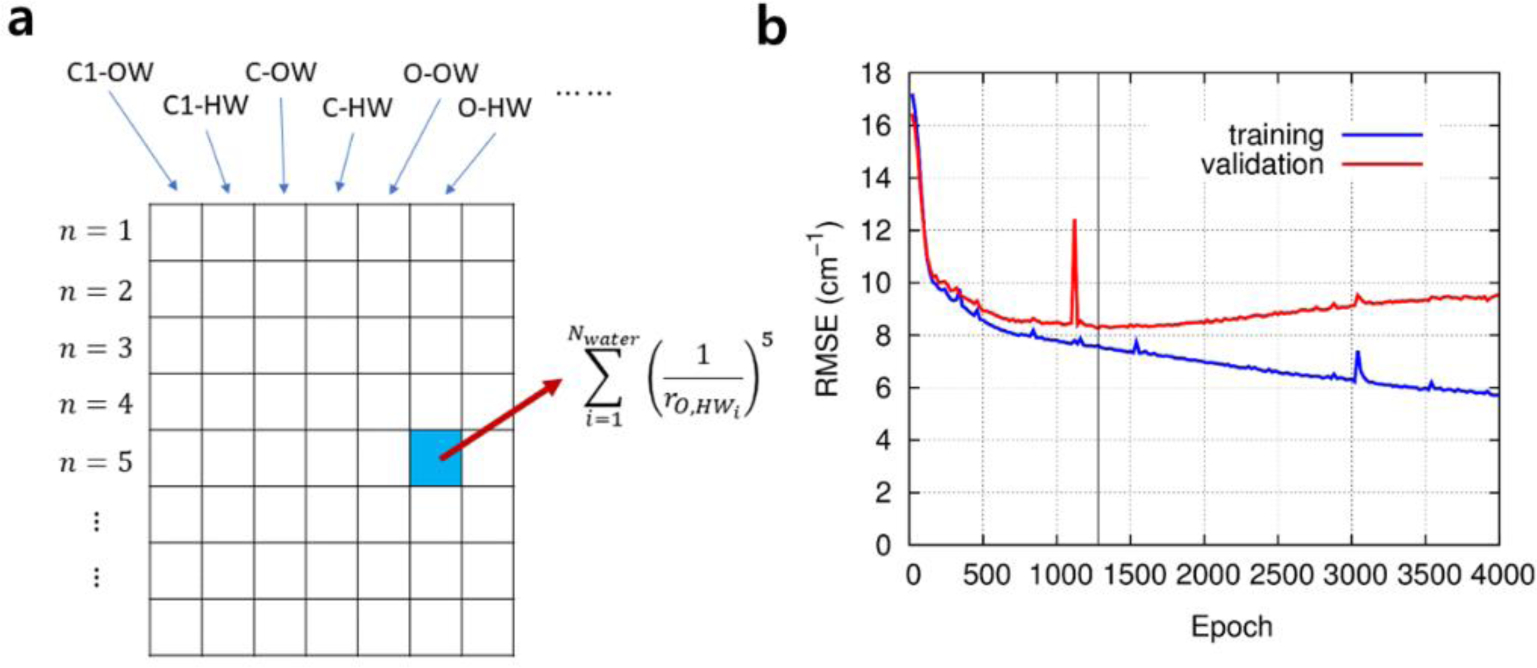
References
-
- Herzberg G Molecular Spectra and Molecular Structure I: Spectra of Diatomic Molecules; Van Nostrand, 1950.
-
- Herzberg G Molecular Spectra and Molecular Structure II: Infrared and Raman of Polyatomic Molecules; Van Nostrand, 1956.
-
- Mukamel S Principles of Nonlinear Optical Spectroscopy; Oxford University Press: Oxford, 1995.
-
- Cho M Two-Dimensional Optical Spectroscopy; CRC Press: Boca Raton, 2009.
-
- Hamm P; Zanni MT Concepts and Methods of 2D Infrared Spectroscopy; Cambridge University Press: New York, 2011.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
