

BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy
- PMID: 32603312
- PMCID: PMC7455078
- DOI: 10.1172/jci.insight.138687
BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy
Abstract
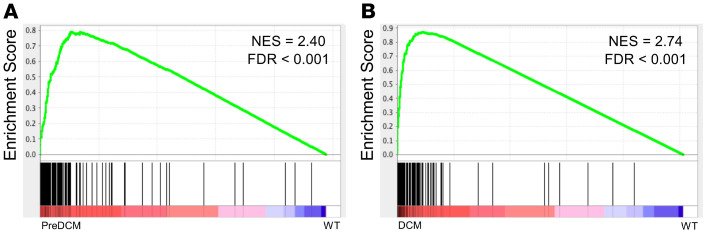
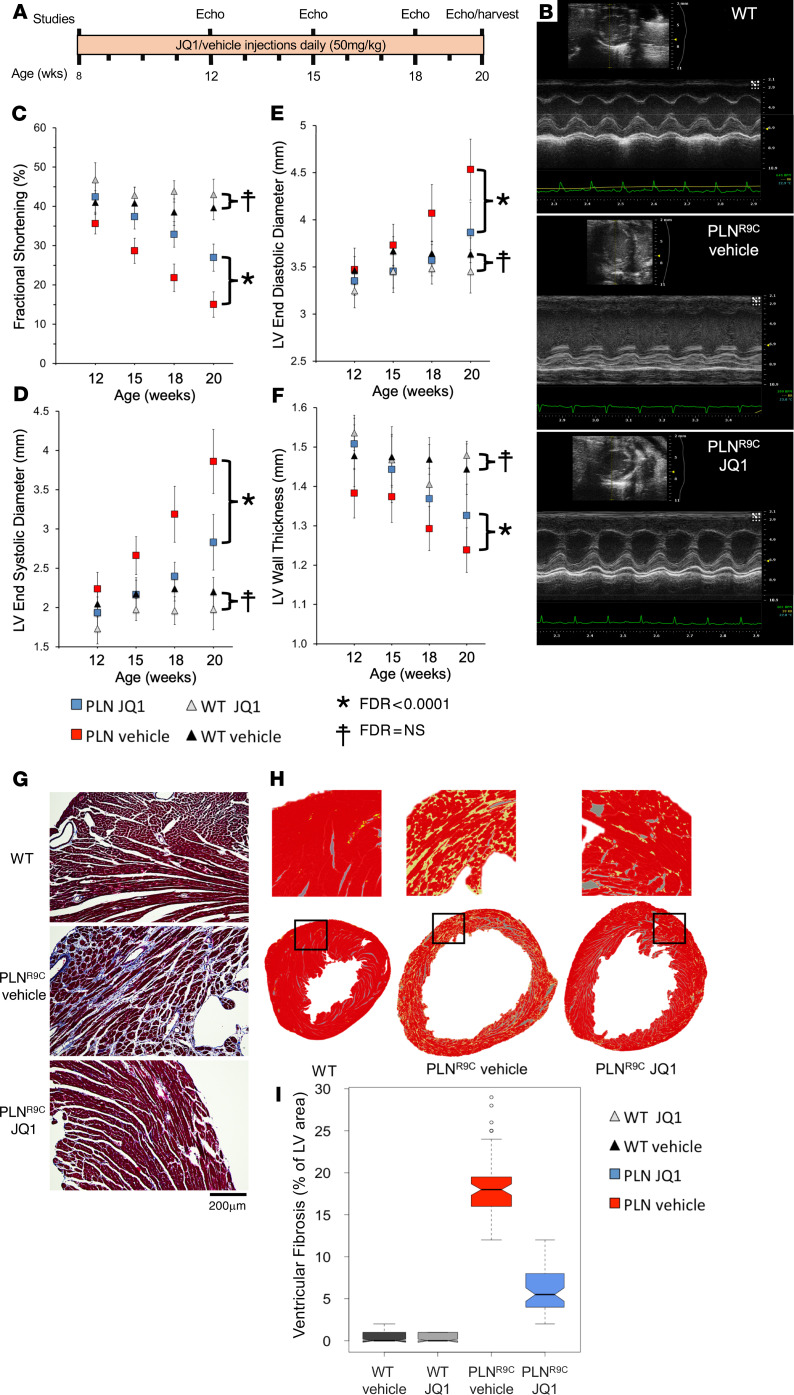
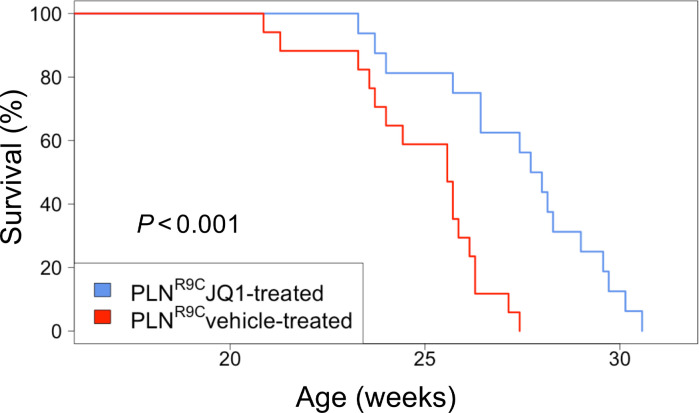
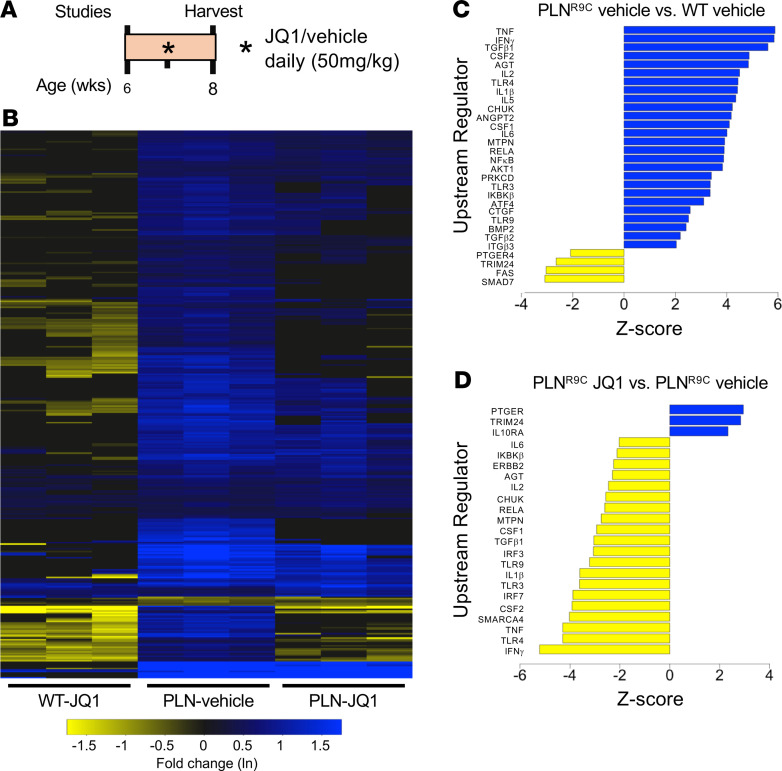
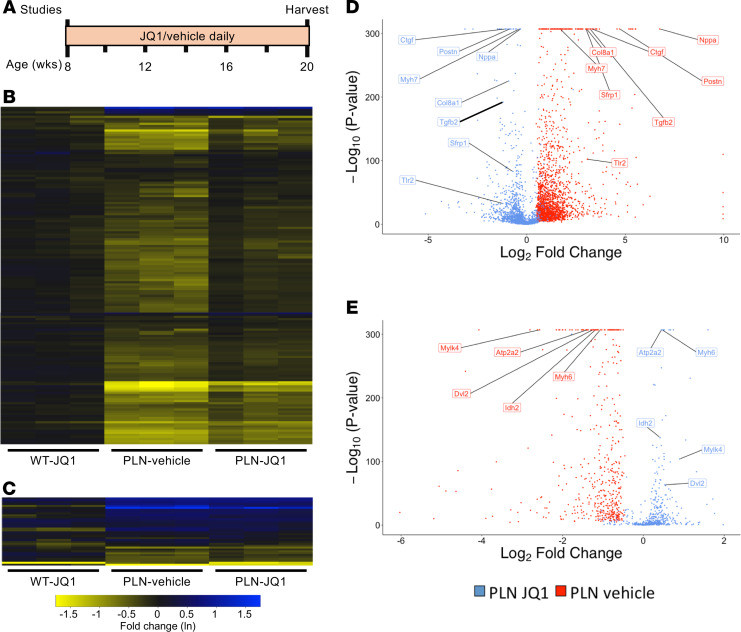
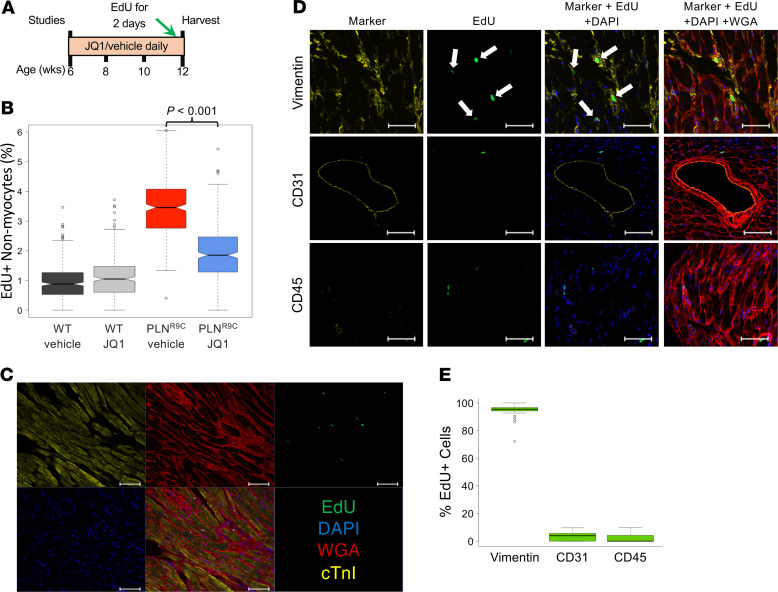
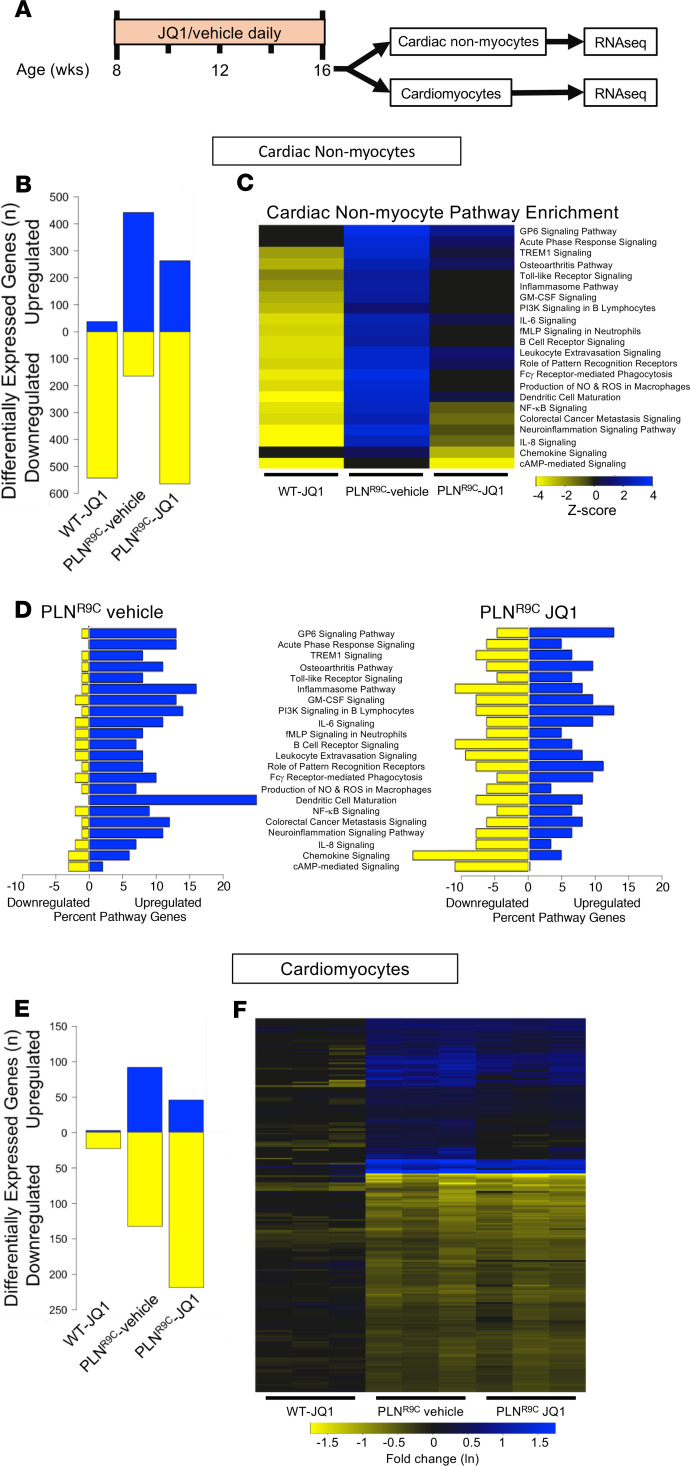
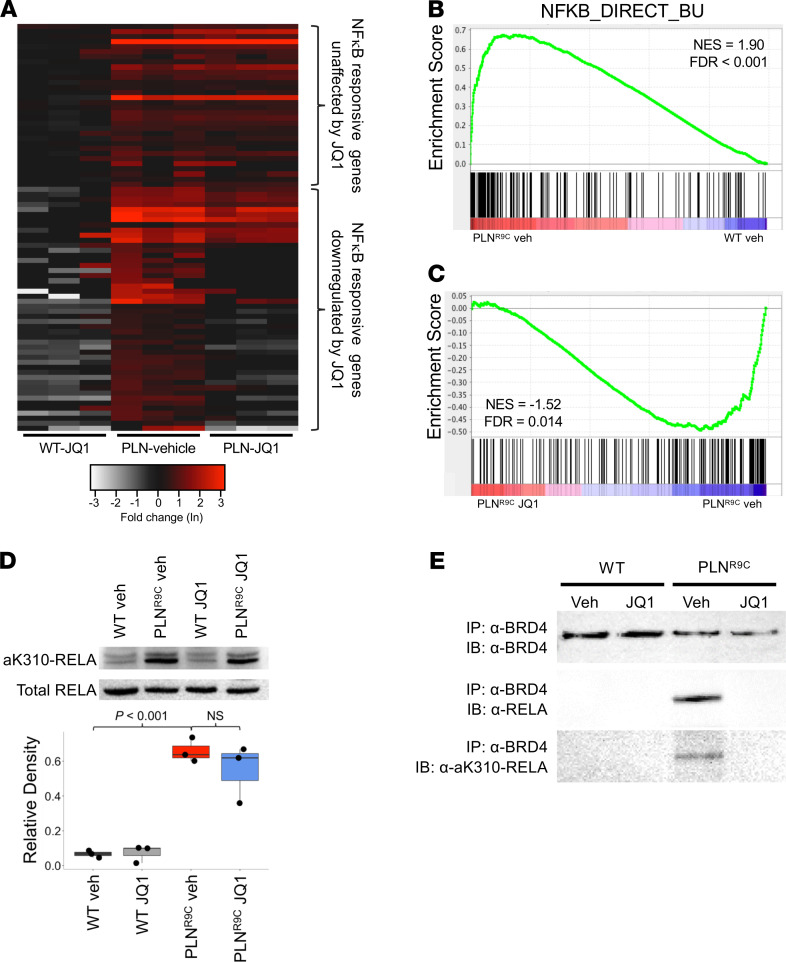
The bromodomain and extraterminal (BET) family comprises epigenetic reader proteins that are important regulators of inflammatory and hypertrophic gene expression in the heart. We previously identified the activation of proinflammatory gene networks as a key early driver of dilated cardiomyopathy (DCM) in transgenic mice expressing a mutant form of phospholamban (PLNR9C) - a genetic cause of DCM in humans. We hypothesized that BETs coactivate this inflammatory process, representing a critical node in the progression of DCM. To test this hypothesis, we treated PLNR9C or age-matched WT mice longitudinally with the small molecule BET bromodomain inhibitor JQ1 or vehicle. BET inhibition abrogated adverse cardiac remodeling, reduced cardiac fibrosis, and prolonged survival in PLNR9C mice by inhibiting expression of proinflammatory gene networks at all stages of disease. Specifically, JQ1 had profound effects on proinflammatory gene network expression in cardiac fibroblasts, while having little effect on gene expression in cardiomyocytes. Cardiac fibroblast proliferation was also substantially reduced by JQ1. Mechanistically, we demonstrated that BRD4 serves as a direct and essential regulator of NF-κB-mediated proinflammatory gene expression in cardiac fibroblasts. Suppressing proinflammatory gene expression via BET bromodomain inhibition could be a novel therapeutic strategy for chronic DCM in humans.
Keywords: Cardiology; Fibrosis; Heart failure; Inflammation.
Conflict of interest statement
Figures
References
-
- Virani SS, et al. Heart disease and stroke statistics — 2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–e596. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
