

Regulation of inflammation in diabetes: From genetics to epigenomics evidence
- PMID: 32603690
- PMCID: PMC7394913
- DOI: 10.1016/j.molmet.2020.101041
Regulation of inflammation in diabetes: From genetics to epigenomics evidence
Abstract
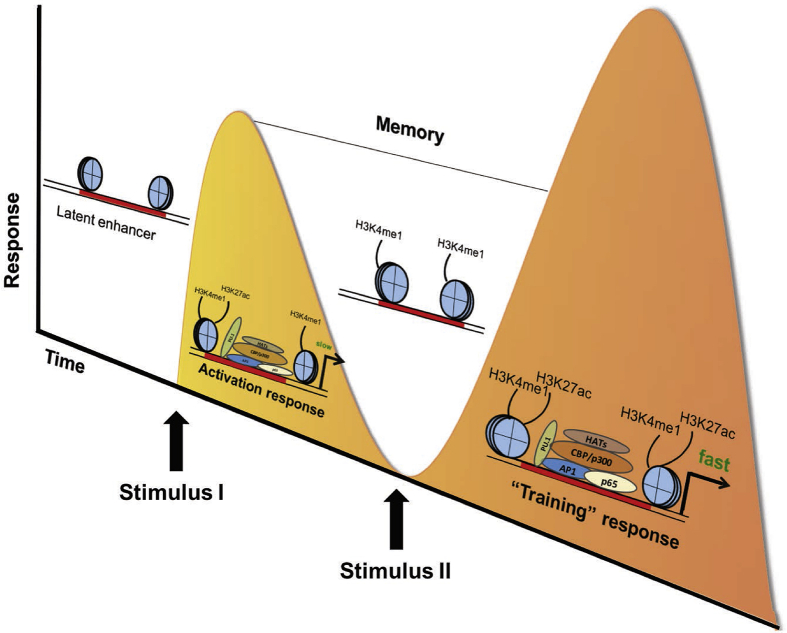
Background: Diabetes is one of the greatest public health challenges worldwide, and we still lack complementary approaches to significantly enhance the efficacy of preventive and therapeutic approaches. Genetic and environmental factors are the culprits involved in diabetes risk. Evidence from the last decade has highlighted that deregulation in the immune and inflammatory responses increase susceptibility to type 1 and type 2 diabetes. Spatiotemporal patterns of gene expression involved in immune cell polarisation depend on genomic enhancer elements in response to inflammatory and metabolic cues. Several studies have reported that most regulatory genetic variants are located in the non-protein coding regions of the genome and particularly in enhancer regions. The progress of high-throughput technologies has permitted the characterisation of enhancer chromatin properties. These advances support the concept that genetic alteration of enhancers may influence the immune and inflammatory responses in relation to diabetes.
Scope of review: Results from genome-wide association studies (GWAS) combined with functional and integrative analyses have elucidated the impacts of some diabetes risk-associated variants that are involved in the regulation of the immune system. Additionally, genetic variant mapping to enhancer regions may alter enhancer status, which in turn leads to aberrant expression of inflammatory genes associated with diabetes susceptibility. The focus of this review was to provide an overview of the current indications that inflammatory processes are regulated at the genetic and epigenomic levels in diabetes, along with perspectives on future research avenues that may improve understanding of the disease.
Major conclusions: In this review, we provide genetic evidence in support of a deregulated immune response as a risk factor in diabetes. We also argue about the importance of enhancer regions in the regulation of immune cell polarisation and how the recent advances using genome-wide methods for enhancer identification have enabled the determination of the impact of enhancer genetic variation on diabetes onset and phenotype. This could eventually lead to better management plans and improved treatment responses in human diabetes.
Keywords: Diabetes; Enhancer; Epigenetics; Genetics; Inflammation.
Copyright © 2020 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures



References
-
- Yamamoto-Honda R., Akanuma Y. vol. 60. World Health Organization; 2002. (Classification of diabetes mellitus). Suppl 7.
-
- Ahlqvist E., Storm P., Käräjämäki A., Martinell M., Dorkhan M., Carlsson A. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. The Lancet Diabetes and Endocrinology. 2018;6(5):361–369. doi: 10.1016/S2213-8587(18)30051-2. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
