

In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes
- PMID: 32605102
- PMCID: PMC7409220
- DOI: 10.3390/microorganisms8070966
In Silico Comparison Shows that the Pan-Genome of a Dairy-Related Bacterial Culture Collection Covers Most Reactions Annotated to Human Microbiomes
Abstract
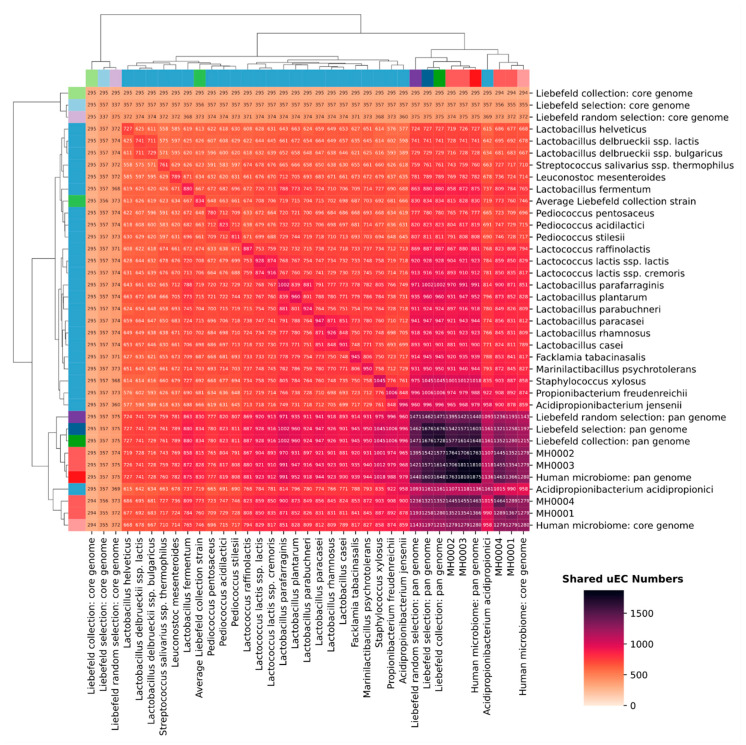
The diversity of the human microbiome is positively associated with human health. However, this diversity is endangered by Westernized dietary patterns that are characterized by a decreased nutrient variety. Diversity might potentially be improved by promoting dietary patterns rich in microbial strains. Various collections of bacterial cultures resulting from a century of dairy research are readily available worldwide, and could be exploited to contribute towards this end. We have conducted a functional in silico analysis of the metagenome of 24 strains, each representing one of the species in a bacterial culture collection composed of 626 sequenced strains, and compared the pathways potentially covered by this metagenome to the intestinal metagenome of four healthy, although overweight, humans. Remarkably, the pan-genome of the 24 strains covers 89% of the human gut microbiome's annotated enzymatic reactions. Furthermore, the dairy microbial collection covers biological pathways, such as methylglyoxal degradation, sulfate reduction, g-aminobutyric (GABA) acid degradation and salicylate degradation, which are differently covered among the four subjects and are involved in a range of cardiometabolic, intestinal, and neurological disorders. We conclude that microbial culture collections derived from dairy research have the genomic potential to complement and restore functional redundancy in human microbiomes.
Keywords: dairy microbiome; diversity; health; human gut microbiome.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
