

The international X-linked hypophosphataemia (XLH) registry (NCT03193476): rationale for and description of an international, observational study
- PMID: 32605590
- PMCID: PMC7329472
- DOI: 10.1186/s13023-020-01434-4
The international X-linked hypophosphataemia (XLH) registry (NCT03193476): rationale for and description of an international, observational study
Abstract
Background: X-linked hypophosphataemia (XLH) is a rare, hereditary, progressive and lifelong phosphate wasting disorder characterised by pathological elevations in fibroblast growth factor (FGF) 23 concentration and activity; XLH has an incidence of approximately 1 in 20-25,000 individuals. Excess FGF23 activity leads to increased phosphate excretion in the kidneys - mediated by downregulation of renal tubular phosphate transporters - and reduced phosphate absorption in the intestines - due to impaired vitamin D activation. This results in impaired bone growth and mineralisation, short and disproportionate stature, leg bowing, musculoskeletal pain, spontaneous dental abscesses, rickets, and osteomalacia. The spectrum of manifestations differs between paediatric and adult patients. Those involved in the treatment of this condition face many challenges, including a lack of robust natural history and demographic data. This multicentre, international, rare-disease patient registry (XLH Registry) was established to address the paucity of data in XLH and to help inform future clinical practice.
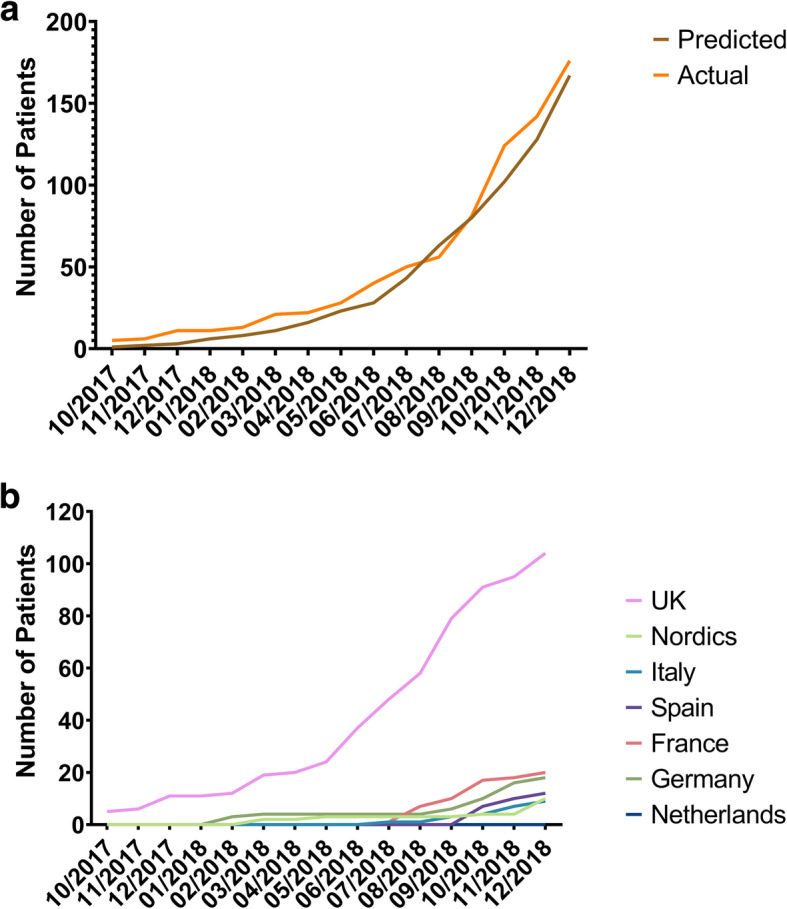
Results: The XLH Registry collects standard diagnostic and monitoring practice data, including (where applicable) diagnosis and disease progression history, treatment regimens and family history; the protocol does not mandate any interventions or clinical assessments. The XLH Registry aims to recruit 1200 paediatric and adult patients with XLH over 10 years, and several data analyses and peer-reviewed publications are expected to be generated throughout this period. A post-authorisation safety study for Bburosumab, for which the registry Sponsor is the marketing authorisation holder, will be nested as a sub-study within the XLH Registry via a subsequent protocol amendment.
Conclusion: The data collected within this rare-disease patient registry will be utilised to synthesise real-world evidence to inform the management of XLH, to improve the quality of life and standard of care of patients living with this rare debilitating disease.
Keywords: Bburosumab; Disease history; Patient registry; Post-authorisation safety; Quality of life; Rare disease; Real-world evidence; Vitamin D; X-linked hypophosphataemia (XLH); XLH management.
Conflict of interest statement
RP received research grant, honorarium as a speaker and travel grant from Kyowa Kirin International.
ON has received research support, speakers’ honoraria, consulting fees from Kyowa Kirin.
OM declares consultancy, travel support and honoraria from Kyowa Kirin.
SSB-N has received fees from Kyowa Kirin for speeches, consultancy, and participation in Advisory Boards. In addition, Kyowa Kirin has provided a pending grant for an upcoming study. Novo Nordisk have paid a fee for an invited speech.
GA declares having received an honorarium for lectures and other educational activities, including travel and registration support for some scientific meetings in the field of Paediatric Nephrology. GA participates as a local investigator at The International X-Linked Hypophosphataemia (XLH) Registry (NCT03193476).
ZM has received honoraria from Kyowa Kirin International for participating in meetings relating to the XLH registry, and payment for lectures on XLH from Kyowa Kirin International.
MS was an employee of the study sponsor between Jan 2017 and July 2019.
EL performed consultancy for Kyowa Kirin.
RJ was engaged as a consultant for the design and implementation of the XLH Registry. RJ has not received remuneration for the authoring of this manuscript.
The other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
