

Identification of cell type-specific methylation signals in bulk whole genome bisulfite sequencing data
- PMID: 32605651
- PMCID: PMC7329512
- DOI: 10.1186/s13059-020-02065-5
Identification of cell type-specific methylation signals in bulk whole genome bisulfite sequencing data
Abstract
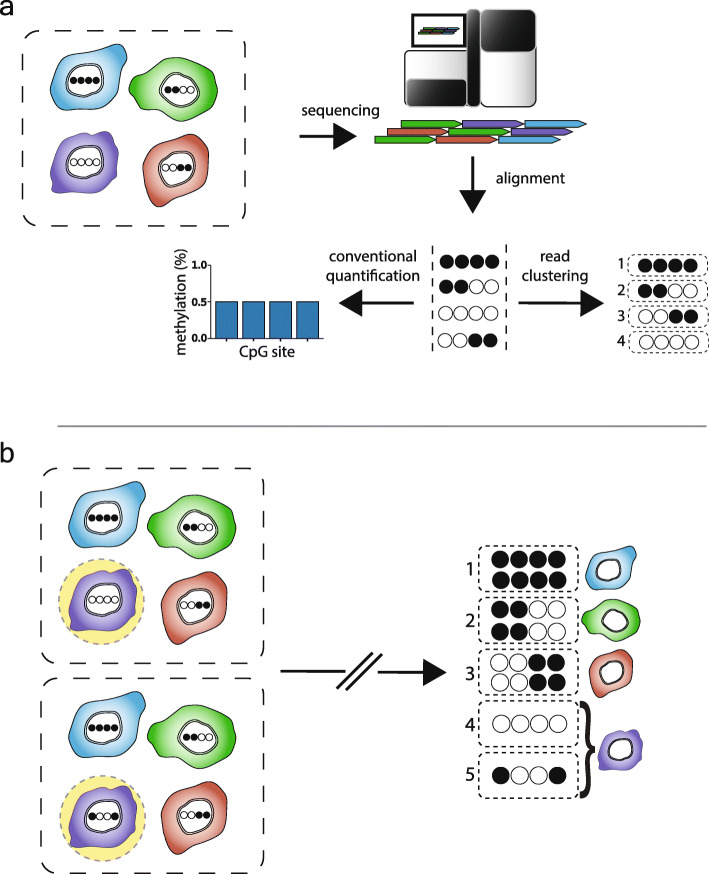
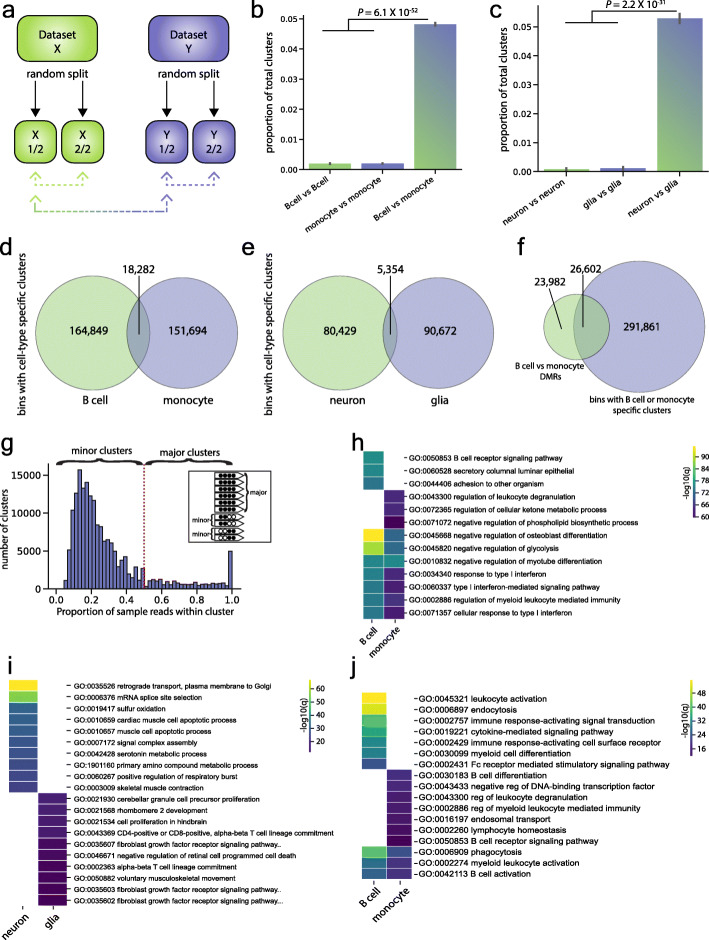
Background: The traditional approach to studying the epigenetic mechanism CpG methylation in tissue samples is to identify regions of concordant differential methylation spanning multiple CpG sites (differentially methylated regions). Variation limited to single or small numbers of CpGs has been assumed to reflect stochastic processes. To test this, we developed software, Cluster-Based analysis of CpG methylation (CluBCpG), and explored variation in read-level CpG methylation patterns in whole genome bisulfite sequencing data.
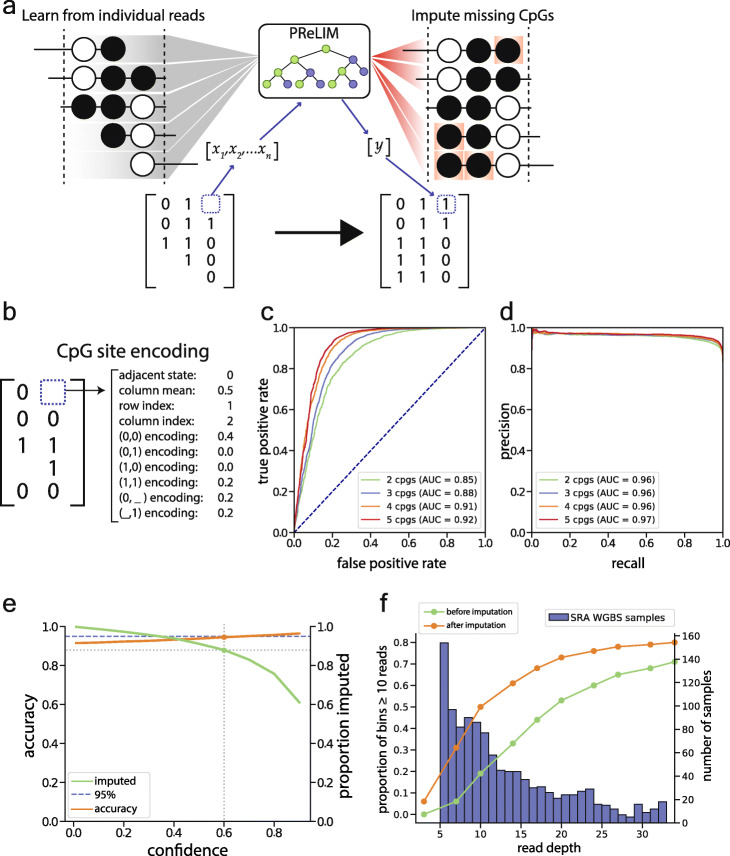
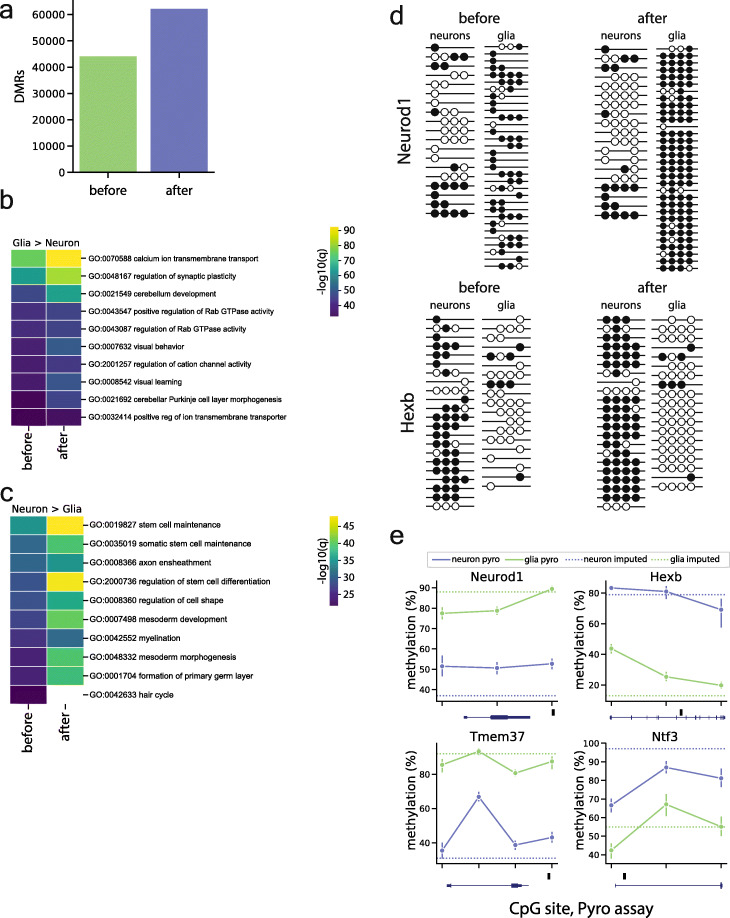
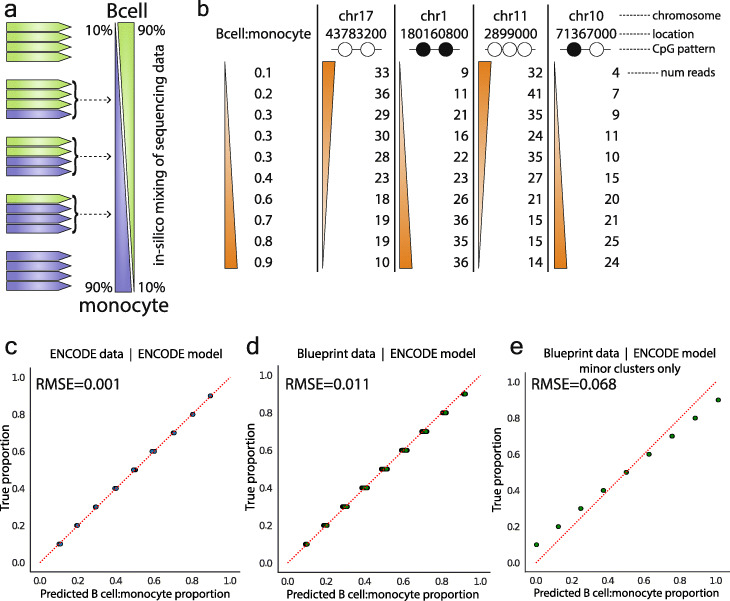
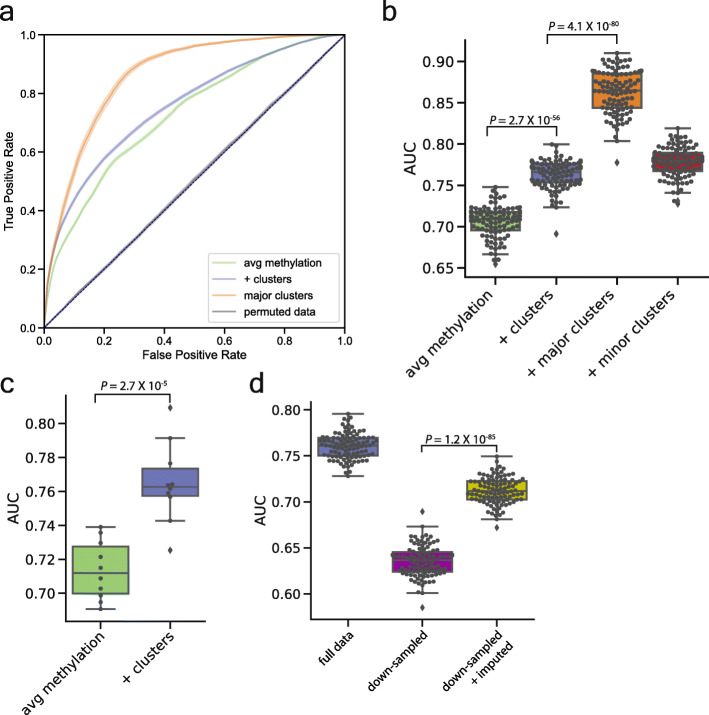
Results: Analysis of both human and mouse whole genome bisulfite sequencing datasets reveals read-level signatures associated with cell type and cell type-specific biological processes. These signatures, which are mostly orthogonal to classical differentially methylated regions, are enriched at cell type-specific enhancers and allow estimation of proportional cell composition in synthetic mixtures and improved prediction of gene expression. In tandem, we developed a machine learning algorithm, Precise Read-Level Imputation of Methylation (PReLIM), to increase coverage of existing whole genome bisulfite sequencing datasets by imputing CpG methylation states on individual sequencing reads. PReLIM both improves CluBCpG coverage and performance and enables identification of novel differentially methylated regions, which we independently validate.
Conclusions: Our data indicate that, rather than stochastic variation, read-level CpG methylation patterns in tissue whole genome bisulfite sequencing libraries reflect cell type. Accordingly, these new computational tools should lead to an improved understanding of epigenetic regulation by DNA methylation.
Keywords: Bisulfite-seq; DNA methylation; Deconvolution; Imputation; Machine learning; Random forests; Read-level; WGBS.
Conflict of interest statement
The authors have no competing interests to report.
Figures
References
-
- Landan G, Cohen NM, Mukamel Z, Bar A, Molchadsky A, Brosh R, Horn-Saban S, Zalcenstein DA, Goldfinger N, Zundelevich A. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat Genet. 2012;44:1207. doi: 10.1038/ng.2442. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
